

Abstract
Leukemia in infants is rare but generates tremendous interest due to its aggressive clinical presentation in a uniquely vulnerable host, its poor response to current therapies, and its fascinating biology. Increasingly, these biological insights are pointing the way toward novel therapeutic approaches. Using representative clinical case presentations, we review the key clinical, pathologic, and epidemiologic features of infant leukemia, including the high frequency of KMT2A gene rearrangements. We describe the current approach to risk-stratified treatment of infant leukemia in the major international cooperative groups. We highlight recent discoveries that elucidate the molecular biology of infant leukemia and suggest novel targeted therapeutic strategies, including modulation of aberrant epigenetic programs, inhibition of signaling pathways, and immunotherapeutics. Finally, we underscore the need for increased global collaboration to translate these discoveries into improved outcomes.
Case studies
Patient 1
A 4-week-old boy was admitted to the hospital with fever, feeding problems, and lethargy. Physical examination revealed hepatosplenomegaly, pallor, and petechiae. Laboratory examination showed striking leukocytosis (white blood cells [WBCs], 1100 × 109/L), anemia (hemoglobin [Hb], 3.1 g/dL), and thrombocytopenia (platelets [PLTs], 10 × 109/L]. Bone marrow examination showed 98% blast cells with a very immature B-cell phenotype (CD19+, CD10−, cytoplasmic immunoglobulin-negative, CD20−) and a KMT2A-MLLT1 gene fusion (Table 1). Cerebrospinal fluid (CSF) examination showed WBCs at 38 × 109/L, and cytospin showed lymphoblasts morphologically. He was enrolled in the Interfant-06 protocol (clinicaltrials.gov NCT00550992). During induction (Figure 1), he had febrile neutropenia and received broad-spectrum empiric antibiotics. He achieved morphological remission after induction.
Nomenclature of infant leukemia genetics
| Gene name | Alias | Chromosome | Translocation | Fusion protein | Alias |
|---|---|---|---|---|---|
| KMT2A | MLL | 11q23 | t(v;11q23) | Variable | |
| Fusion partners (selected) | |||||
| AFF1 | AF4 | 4q21 | t(4;11)(q21;q23) | KMT2A-AFF1 | MLL-AF4 |
| MLLT1 | ENL | 19p13.3 | t(11;19)(q23;p13.3) | KMT2A-MLLT1 | MLL-ENL |
| MLLT3 | AF9 | 9p21 | t(9;11)(p21;q23) | KMT2A-MLLT3 | MLL-AF9 |
| MLLT10 | AF10 | 10p12 | t(10;11)(p12;q23) | KMT2A-MLLT10 | MLL-AF10 |
| ELL | — | 19p13.1 | t(11;19)(q23;p13.1) | KMT2A-ELL | MLL-ELL |
| Gene name | Alias | Chromosome | Translocation | Fusion protein | Alias |
|---|---|---|---|---|---|
| KMT2A | MLL | 11q23 | t(v;11q23) | Variable | |
| Fusion partners (selected) | |||||
| AFF1 | AF4 | 4q21 | t(4;11)(q21;q23) | KMT2A-AFF1 | MLL-AF4 |
| MLLT1 | ENL | 19p13.3 | t(11;19)(q23;p13.3) | KMT2A-MLLT1 | MLL-ENL |
| MLLT3 | AF9 | 9p21 | t(9;11)(p21;q23) | KMT2A-MLLT3 | MLL-AF9 |
| MLLT10 | AF10 | 10p12 | t(10;11)(p12;q23) | KMT2A-MLLT10 | MLL-AF10 |
| ELL | — | 19p13.1 | t(11;19)(q23;p13.1) | KMT2A-ELL | MLL-ELL |
—, no alias; v, variable chromosome.
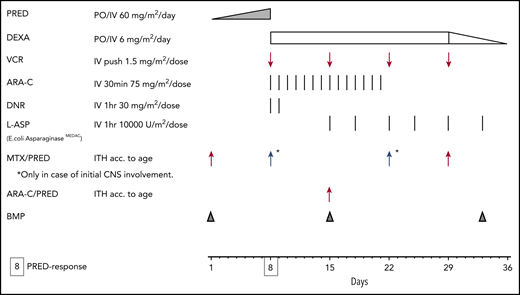
Interfant induction, now used by all 3 cooperative groups (Interfant, COG, and JPLSG). ARA-C, cytarabine; BMP, bone marrow procedure; DEXA, dexamethasone; DNR, daunorubicin; ITH, intrathecal; MTX, methotrexate; PRED, prednisone; VCR, vincristine.
Interfant induction, now used by all 3 cooperative groups (Interfant, COG, and JPLSG). ARA-C, cytarabine; BMP, bone marrow procedure; DEXA, dexamethasone; DNR, daunorubicin; ITH, intrathecal; MTX, methotrexate; PRED, prednisone; VCR, vincristine.
Patient 2
A 4-month-old boy was admitted to the hospital because he was not feeding well and had pale gray skin. Physical examination revealed pallor and hepatosplenomegaly. Laboratory examination showed leukocytosis (WBCs, 250 × 109/L), anemia (Hb, 5.5 g/dL), and thrombocytopenia (PLTs, 28 × 109/L). Bone marrow examination showed 99% blast cells with very immature B-cell phenotype (CD19+, CD10−, cytoplasmic immunoglobulin-negative, CD20−) and a KMT2A-AFF1 gene fusion. CSF examination showed WBCs at 3 × 109/L, and cytospin showed lymphoblasts morphologically. He was enrolled in the Children’s Oncology Group (COG) protocol AALL0631 (clinicaltrials.gov NCT00557193). During induction, he had febrile neutropenia and received broad-spectrum empiric antibiotics. He achieved morphological remission after induction.
Patient 3
A 10-month-old girl was admitted to the hospital with irritability, fever, poor appetite, and rash. Physical examination revealed petechiae, pallor, and tenderness to palpation of the extremities. Laboratory examination showed leukopenia and neutropenia (WBCs, 1.2 × 109/L with 30% neutrophils), anemia (Hb, 6.7 g/dL), and thrombocytopenia (PLTs, 47 × 109/L). Bone marrow examination showed 99% blast cells with immature B-cell phenotype (CD19+, CD10+, cytoplasmic immunoglobulin-negative, CD20−) and normal cytogenetics. CSF examination showed WBCs at 1 × 109/L, and cytospin showed no lymphoblasts morphologically. She was enrolled in the Interfant-06 protocol. She had no significant complications during induction, and achieved morphological remission after induction.
Patient 4
A 7-month-old girl was admitted to the hospital with a progressive rash, fever, and poor feeding. Physical examination revealed scattered palpable blue cutaneous nodules and hepatosplenomegaly. Laboratory examination showed leukocytosis (WBCs, 42 × 109/L), anemia (Hb, 5.9 g/dL), and thrombocytopenia (PLTs, 28 × 109/L). Bone marrow examination showed 78% blast cells with a monoblastic phenotype (MPO+, CD34−, HLA-DR+, CD33+, CD11b+) and KMT2A-MLLT3 fusion. CSF examination showed WBCs at 22 × 109/L, and cytospin showed monoblasts morphologically. Skin biopsy showed leukemia cutis. She was enrolled in the COG AAML1031 protocol (clinicaltrials.gov NCT01371981). During induction with cytarabine, daunorubicin, and etoposide (ADE), she developed gram-negative sepsis, but fully recovered. She achieved morphological remission after induction, including resolution of leukemia cutis.
Patient 5
A 4-month-old boy was admitted with poor feeding, pale skin, and enlarging abdomen. Physical examination revealed pallor and hepatosplenomegaly. Laboratory examination showed leukopenia and neutropenia (WBCs, 2.1 × 109/L with 25% neutrophils), anemia (Hb, 5.4 g/dL), and thrombocytopenia (PLTs, 56 × 109/L). Bone marrow was difficult to aspirate, but sufficient material was obtained for flow cytometry, showing 23% blast cells with a megakaryoblastic phenotype (myeloperoxidase-negative [MPO−], CD41a+, CD61+) and RBM15-MKL1 fusion. Bone marrow biopsy showed striking fibrosis. CSF examination showed WBCs at 0 × 109/L, and cytospin showed no blasts morphologically. He was enrolled in the COG AAML1031 protocol. During induction, he developed a presumed fungal pneumonia (nodular infiltrates on computed tomography with a positive serum galactomannan) that resolved with liposomal amphotericin B. The end-induction bone marrow showed 15% residual megakaryoblasts.
Patient 6
A 7-month-old girl was admitted to the hospital because of a worsening rash, fever, and poor feeding. Physical examination revealed petechiae, pallor, and diffuse adenopathy. Laboratory examination showed leukocytosis (WBCs, 95 × 109/L), anemia (Hb, 6.5 g/dL), and thrombocytopenia (PLTs, 14 × 109/L). Bone marrow examination showed 94% blast cells with 2 discrete populations of blasts: ∼60% of the blasts had an immature B-cell phenotype (CD19+, CD10−, cytoplasmic immunoglobulin-negative, CD20−) and ∼40% had a monoblastic phenotype (MPO+, CD34−, HLA-DR+, CD33+, CD11b+). In addition, a KMT2A-MLLT10 gene fusion was detected. CSF examination showed WBCs at 13 × 109/L, and cytospin showed heterogeneous blasts morphologically. She was treated with induction according to the Interfant-06 protocol. During induction, she had febrile neutropenia and received broad-spectrum empiric antibiotics. She achieved morphological remission after induction.
A summary of the 6 patient cases is provided in Table 2.
Summary of patient cases
| Patient | Diagnostic category | Age, mo/Sex | Molecular abnormality | Induction | Complications | Response |
|---|---|---|---|---|---|---|
| 1 | High-risk KMT2A-rearranged B-ALL | 1/M | KMT2A-MLLT1 | Interfant | Febrile neutropenia | Remission |
| 2 | Intermediate-risk KMT2A-rearranged B-ALL | 4/M | KMT2A-AFF1 | Interfant | Febrile neutropenia | Remission |
| 3 | Low-risk wt-KMT2A B-ALL | 10/F | None | Interfant | None | Remission |
| 4 | KMT2A-rearranged AML | 7/F | KMT2A-MLLT3 | ADE* | Gram-negative sepsis | Remission |
| 5 | wt-KMT2A megakaryoblastic AML | 4/M | RBM15-MKL1 | ADE* | Fungal pneumonia | Induction failure |
| 6 | KMT2A-rearranged MPAL (B/myeloid) | 7/F | KMT2A-MLLT10 | Interfant | Febrile neutropenia | Remission |
| Patient | Diagnostic category | Age, mo/Sex | Molecular abnormality | Induction | Complications | Response |
|---|---|---|---|---|---|---|
| 1 | High-risk KMT2A-rearranged B-ALL | 1/M | KMT2A-MLLT1 | Interfant | Febrile neutropenia | Remission |
| 2 | Intermediate-risk KMT2A-rearranged B-ALL | 4/M | KMT2A-AFF1 | Interfant | Febrile neutropenia | Remission |
| 3 | Low-risk wt-KMT2A B-ALL | 10/F | None | Interfant | None | Remission |
| 4 | KMT2A-rearranged AML | 7/F | KMT2A-MLLT3 | ADE* | Gram-negative sepsis | Remission |
| 5 | wt-KMT2A megakaryoblastic AML | 4/M | RBM15-MKL1 | ADE* | Fungal pneumonia | Induction failure |
| 6 | KMT2A-rearranged MPAL (B/myeloid) | 7/F | KMT2A-MLLT10 | Interfant | Febrile neutropenia | Remission |
F, female; Interfant, Interfant induction (Figure 1); M, male; MPAL, mixed phenotype acute leukemia.
ADE indicates cytarabine 100 mg/m2 every 12 hours days 1 to 10; daunorubicin 50 mg/m2 days 1, 3, and 5; etoposide 100 mg/m2 days 1 to 5.
Discussion: part 1
Definition and epidemiology
“Infant leukemia” refers to acute leukemia diagnosed prior to 1 year of age. Fortunately, infant leukemia is rare, with estimated incidence of 41 cases per million in the United States, which equates to ∼160 cases of infant leukemia per year. Within the infant age group, neuroblastoma, brain tumors, and acute leukemia all occur with similar frequency. There is a slight predominance of lymphoid over myeloid cases within infant leukemia, and of the lymphoid cases, nearly all are B-lineage with <5% T-lineage. The incidence of acute lymphoblastic leukemia (ALL) in infants is lower than in children aged 1 to 14 years old and approximately the same as the incidence of ALL in adolescents. In contrast, the incidence of acute myeloid leukemia (AML) in infants is about twice that of the incidence of AML in older children and adolescents. Interestingly, infant leukemia demonstrates a female predominance, in contrast to the male predominance in leukemia diagnosed beyond the first birthday.1
Clinical features
Prognosis
The prognostic significance of infant age differs between ALL and AML. In ALL, infants fare far worse than older children. The 4-year event-free survival (EFS) in Interfant-99, the largest trial of infant ALL to date, was 47%.4 Recent trials for childhood ALL report long-term EFS rates exceeding 85%.5,6 In AML, outcomes for infants are similar to those for older children.3
KMT2A (formerly MLL) gene rearrangements
A high proportion of infant leukemias are characterized cytogenetically by balanced chromosomal translocations involving the histone lysine methyltransferase 2A gene (KMT2A, formerly known as mixed lineage leukemia [MLL] gene) at chromosome 11q23. KMT2A rearrangements (KMT2A-r) occur in ∼5% of childhood ALL cases overall,7 but in 70% to 80% of ALL in infants.2,4 In childhood AML, KMT2A-r is more common overall (15%-20%), but is also particularly common in the infant age group (∼50%).8
KMT2A-r results in the fusion of the N terminus of the KMT2A gene with the C terminus of a partner gene. Remarkably, 94 different KMT2A partner genes have now been identified.9 In infant ALL, 4 partner genes account for 93% of cases: AFF1 (formerly AF4; 49%), MLLT1 (formerly ENL; 22%), MLLT3 (formerly AF9; 17%), and MLLT10 (formerly AF10; 5%). In infant AML, 3 partner genes account for 66% of cases: MLLT3 (22%), MLLT10 (27%), and ELL (17%).
Various lines of evidence (eg, retrospective analyses of neonatal samples10 and twin concordance studies11 ) have shown that KMT2A rearrangements are acquired in hematopoietic precursors in utero, and, compared with other oncogenic fusions such as ETV6-RUNX1, initiate a strikingly rapid progression to leukemia. One intriguing aspect of leukemia epidemiology is that KMT2A-r leukemias occur with high frequency in 2 very different clinical situations: (1) infants with de novo acute leukemia and (2) patients with treatment-related secondary myelodysplastic syndrome/AML after exposure to potent DNA topoisomerase II (DNAt2) inhibitors (eg, etoposide). This has led to a hypothesis, with supporting evidence from case-control12,13 and laboratory14 studies, that maternal exposure to environmental DNAt2 inhibitors (eg, dietary flavonoids) during pregnancy may contribute to the risk of KMT2A-r infant leukemia. Germline genetic susceptibility may also play a role, as candidate gene studies15,16 and genome-wide association studies17,18 have identified a number of single-nucleotide polymorphisms that correlate with risk of infant leukemia.
In ALL, KMT2A-r is associated with CD10 negativity and coexpression of 1 or more myeloid antigens, suggesting that these leukemias arise from very immature lymphoid progenitors.19 In AML, KMT2A-r is associated with monocytic differentiation.3 Infant leukemia cases can be of ambiguous lineage, either due to a mixed phenotype (mixed phenotype acute leukemia [MPAL], as shown in patient 6) or to lack of differentiation markers (acute undifferentiated leukemia).
KMT2A-r has different prognostic implications in infant ALL than infant AML. In infant ALL, KMT2A-r is clearly associated with poorer outcome. In the Children’s Cancer Group protocol CCG-1953, 5-year EFS for KMT2A-r infants was 34% vs 60% with germline (wild type) KMT2A (wt-KMT2A).2 In Interfant-99, 4-year EFS in KMT2A-r and wt-KMT2A infants was 37% and 74%, respectively.4 In infant AML, KMT2A-r is not a significant risk factor. In a combined analysis of AML-BFM-98 and -2004, the 5-year EFS was 43% and 52% for KMT2A-r and wt-KMT2A infants, respectively (P = .59).3 There is, however, evidence in pediatric AML generally that within KMT2A-r cases, certain KMT2A fusion partners may be associated with favorable (eg, MLLT11) or unfavorable (eg, MLLT10) prognosis, and there are plans to incorporate these into future pediatric AML risk-stratification algorithms.20,21
Among infants with KMT2A-r ALL, additional independent prognostic factors include age and WBCs at diagnosis, with the younger infants and those with the higher WBCs having poorer outcomes.2,4,22 In the context of a 7-day “prophase” of single-agent prednisone given prior to intensive induction chemotherapy in the Interfant-99 protocol, a poor response (≥1000 blasts per microliter in the peripheral blood on day 8) was also an independent negative prognostic factor.4
Induction therapy and risk stratification: infant AML
Given the similar prognosis and response to therapy of infants with AML compared with older children, infants are generally treated on the same clinical trial protocols as older children, which typically include intensive multiagent chemotherapy to induce remission followed by consolidation with either additional chemotherapy courses (for patients with favorable prognostic features) or allogeneic hematopoietic stem cell transplantation (HSCT; for patients with unfavorable prognostic features). Gemtuzumab is likely to be added to chemotherapy in future protocols based on favorable results, both in pediatrics overall23 and infants specifically.24 Cases of AML in infants are relatively unlikely to harbor cytogenetic or molecular abnormalities that confer unfavorable risk or favorable risk, and risk stratification is typically dictated by end-induction minimal residual disease (MRD) testing, although the KMT2A fusion partner may be incorporated in future protocols.
One notable exception to this, however, is illustrated in patient 5. Among pediatric cases of acute megakaryoblastic leukemia, the vast majority occur in patients with Down syndrome (constitutional trisomy 21), typically characterized by presentation at age 2 to 4 years, a history of transient myeloproliferative disorder in the newborn period, and a favorable prognosis.25 When acute megakaryoblastic leukemia occurs in children without Down syndrome, on the other hand, the patient is nearly always an infant. A variety of oncogenic fusions, of which the first to be described was the RBM15-MKL1 fusion resulting from the reciprocal translocation t(1;22)(p13;q13), drive this rare subset of infant leukemia. The prognosis is generally unfavorable due to a high risk of chemotherapy resistance and relapse, although outcomes do vary according to the specific oncogenic fusion.26
Induction therapy and risk stratification: infant ALL
Treatment of infant ALL is quite different than that for childhood ALL generally. There are 3 major cooperative groups conducting specific clinical trials for infant ALL: Interfant (based in Europe), COG (based in North America), and the Japanese Pediatric Leukemia Study Group (JPLSG). All have adopted an identical induction strategy based on Interfant-99 (Figure 1).4 Until recently, COG trials used a more intensive induction regimen, but this was abandoned in favor of the Interfant induction due to excessive toxicity.27 In recently completed trials, all used a prospective risk-stratified approach that incorporates KMT2A-r status and age (Table 3).
Risk stratification of infant ALL
| Risk | Interfant | COG | JPLSG | Approximate EFS, % |
|---|---|---|---|---|
| High | KMT2A-r and age <6 mo and WBCs ≥ 300 000/μL | KMT2A-r and age <3 mo | KMT2A-r and (age <6 mo or CNS leukemia) | 20 |
| Intermediate | KMT2A-r and not high risk | KMT2A-r and not high risk | KMT2A-r and not high risk | 50 |
| Low | wt-KMT2A | wt-KMT2A | wt-KMT2A | 75 |
| Risk | Interfant | COG | JPLSG | Approximate EFS, % |
|---|---|---|---|---|
| High | KMT2A-r and age <6 mo and WBCs ≥ 300 000/μL | KMT2A-r and age <3 mo | KMT2A-r and (age <6 mo or CNS leukemia) | 20 |
| Intermediate | KMT2A-r and not high risk | KMT2A-r and not high risk | KMT2A-r and not high risk | 50 |
| Low | wt-KMT2A | wt-KMT2A | wt-KMT2A | 75 |
Among infant ALL patients, wt-KMT2A patients are considered low risk, and have favorable clinical features (eg, lower WBCs, older age at presentation). However, the outcome of wt-KMT2A infants is clearly inferior to ALL patients diagnosed after 1 year of age. This is likely due, in part, to differences in distribution of favorable genetic features, as infants with wt-KMT2A ALL are much less likely to harbor the favorable genetic features high hyperdiploidy and ETV6–RUNX1 fusions compared with older children.28,29 Moving forward, wt-KMT2A infant ALL patients are likely to be treated on general frontline ALL protocols, and therefore subject to the same risk-group stratification strategies, including the prospective use of MRD monitoring.
Induction-therapy complications
The infant patient’s unique vulnerability to complications and toxicities presents a challenge in treating infant acute leukemia. There are limited data to guide how the distinct and rapidly changing physiology of infants (in terms of body composition, binding of drugs by plasma proteins, cytochrome p450 activity, renal function, immunocompetence, etc) should be considered in designing chemotherapy treatment protocols. The data that do exist differ by chemotherapy drug. For example, no age dependency was found for the pharmacokinetics of daunorubicin30 whereas the systemic clearance rate of methotrexate tended to increase with age during infancy.31 Infant leukemia protocols have encountered problems with excessive toxicity, both in ALL27 and AML.32 Survivors of infant leukemia also demonstrate an increased risk of late effects, particularly in cases where treatment included cranial radiation or HSCT.25
Case studies, continued
Patient 1
Postinduction, patient 1 was randomized to receive consolidation with multiagent chemotherapy blocks protocol IB and MARMA. His MRD level post-MARMA was <10e−4 (0.01%) by polymerase chain reaction. He received allogeneic HSCT in first remission with an umbilical cord blood donor after a preparatory regimen with busulfan, cyclophosphamide, and melphalan. He relapsed 7 months after HSCT. His leukemia was refractory to several attempts at reinduction with various multiagent salvage chemotherapy regimens and he died of overwhelming sepsis and multiorgan failure at age 17 months.
Patient 2
Postinduction, patient 2 was randomized to receive consolidation with multiagent chemotherapy blocks (reinduction, consolidation, continuation) without the FLT3 inhibitor lestaurtinib, followed by maintenance therapy for a total duration of 2 years. He is now 5 years after initial diagnosis and remains in complete remission.
Patient 3
Postinduction, patient 3 received multiagent chemotherapy blocks protocol IB, MARMA, and OCTADAD, followed by maintenance for a total duration of 2 years. She is now 7 years after initial diagnosis and remains in complete remission.
Patient 4
Postinduction, patient 4 received 3 additional blocks of intensive multiagent chemotherapy. During recovery from the third and final postinduction block, she developed a rash that was biopsied and shown to represent relapsed leukemia cutis. Bone marrow and CSF also revealed recurrent disease (8% blasts in the bone marrow, WBCs at 7 × 109/L in CSF with blasts on cytospin). She received a salvage regimen with azacitidine and fludarabine, cytarabine, and granulocyte colony-stimulating factor (FLAG) chemotherapy. She achieved a second remission and proceeded to allogeneic HSCT with an HLA-haploidentical bone marrow donor after a preparatory regimen with busulfan and cyclophosphamide. She is now 2 years post-HSCT and remains in second remission.
Patient 5
Because patient 5 was refractory to induction therapy, he received a salvage chemotherapy regimen with mitoxantrone and high-dose cytarabine. He achieved a morphologic remission, but still had scattered detectable megakaryoblasts by morphology and 3% megakaryoblasts by flow cytometry. He received allogeneic HSCT with an umbilical cord blood donor after a preparatory regimen with busulfan, cyclophosphamide, and melphalan. He died of multiorgan failure in the setting of progressive fungal infection 4 months after HSCT. At autopsy, residual leukemia was seen in the bone marrow.
Patient 6
For patient 6, postinduction, flow cytometry revealed a residual population of 2.8% in the bone marrow that expressed the same monoblastic phenotype (MPO+, CD34−, HLA-DR+, CD33+, CD11b+) that was detected as the minority population at diagnosis. She received a second round of induction therapy with ADE, complicated by Streptococcus viridans sepsis. She recovered and was in complete remission with negative flow cytometry. She received allogeneic HSCT with an HLA-identical sibling bone marrow donor after a preparatory regimen with busulfan and cyclophosphamide. She remains in remission 3 years after HSCT.
These cases are summarized in Table 2.
Discussion: part 2
Postinduction therapy for infant leukemia
Postinduction management of infant leukemia is heterogeneous. Table 4 summarizes the postinduction treatment approaches for infant ALL by the major cooperative groups (see also, Table 3). Interfant-06 tested whether consolidation with “myeloid”-style chemotherapy with cytarabine, daunorubicin, mitoxantrone, and etoposide is superior to “lymphoid”-style consolidation with cyclophosphamide, cytarabine and 6-mercaptopurine in KMT2A-r infants. This stems from the hypothesis that these leukemias derive from an early hematopoietic precursor with myeloid differentiation potential and may therefore respond better to chemotherapy regimens developed for AML. AALL0631 tested whether the addition of an FLT3 tyrosine kinase inhibitor (lestaurtinib) to postinduction chemotherapy will enhance the effectiveness of chemotherapy, based on data showing aberrant activation of the FLT3 pathway in KMT2A-r ALL. Given the similarities in treatment approach and outcomes between the groups and the rarity of infant ALL, the 3 groups are currently developing a joint collaborative protocol to standardize treatment and enhance the ability to test novel treatment approaches based on recent discoveries regarding the unique molecular biology of KMT2A-r leukemias and ongoing pilot trials within the groups (detailed in subsequent sections).
Postinduction approaches for infant ALL in 3 major cooperative groups on most recent trials
| Interfant* | COG | JPLSG | |
|---|---|---|---|
| Trial | Interfant-06 | AALL0631 | MLL-10 |
| Randomized postinduction intervention | Protocol IB vs ADE/MAE | ±Lestaurtinib (FLT3 tyrosine kinase inhibitor) | None (single arm) |
| HSCT | All high risk, plus MRD+ after MARMA | None | All high risk |
| Interfant* | COG | JPLSG | |
|---|---|---|---|
| Trial | Interfant-06 | AALL0631 | MLL-10 |
| Randomized postinduction intervention | Protocol IB vs ADE/MAE | ±Lestaurtinib (FLT3 tyrosine kinase inhibitor) | None (single arm) |
| HSCT | All high risk, plus MRD+ after MARMA | None | All high risk |
Protocol IB consolidation indicates cyclophosphamide 1000 mg/m2 days 1 and 29; cytarabine 75 mg/m2 days 3 to 6, 10 to 13, 17 to 20, 24 to 27; 6-mercaptopurine 60 mg/m2 days 1 to 28 consolidation. ADE/MAE consolidation indicates cytarabine 100 mg/m2 every 12 hours days 1 to 10; daunorubicin 50 mg/m2 days 1, 3, and 5; etoposide 100 mg/m2 days 1 to 5/mitoxantrone 12 mg/m2 days 1, 3, and 5; cytarabine 100 mg/m2 every 12 hours days 1 to 10; etoposide 100 mg/m2 days 1 to 5. MARMA indicates methotrexate 5000 mg/m2 days 1 and 8; 6-mercaptopurine 25 mg/m2 days 1 to 14; cytarabine 3000 mg/m2 every 12 hours days 15, 16, 22 and 23; pegylated asparaginase 2500 IU/m2 day 23.
Postinduction treatment of infant AML is typically the same as for older children with AML. Patient 6 demonstrates a typical induction and postinduction course for an infant with MPAL. The decision of whether to proceed with induction treatment following an ALL or AML protocol is generally made based on whether the lymphoid or myeloid lineage appears to be predominant. Postinduction treatment should factor in the response to the chosen induction therapy.
HSCT in infant leukemia
The use of HSCT in infant leukemia is variable, reflecting uncertainty regarding the risk/benefit ratio of HSCT in this population. The published data (concisely reviewed by Sison and Brown33 ) are not conclusive. There does appear to be a small minority of KMT2A-r patients at high risk of relapse (very young age, very high WBCs, and persistence of MRD) who may benefit from HSCT in first remission.34,35
Chemotherapy resistance and relapse in infant leukemia
Poor initial response to prednisone is significantly more common in infants than in older children with ALL, and infant KMT2A-r ALL cells demonstrate enhanced in vitro resistance to corticosteroids and asparaginase in assays using short-term exposure of bulk leukemia populations. Conversely, these cells appear to be particularly sensitive to nucleoside analogs like cytarabine, perhaps related to high expression levels of the membrane-bound nucleoside transporter ENT1.36,37 Infant KMT2A-r AML cells do not demonstrate enhanced resistance or sensitivity.38-40
Clinical data do not support the hypothesis that bulk leukemia chemoresistance is responsible for poor outcomes. The typical pattern of failure for infant KMT2A-r ALL is to achieve rapid complete remission with induction chemotherapy, but then relapse several months later during active therapy. This suggests that the poor outcomes are due primarily to the emergence of a chemoresistant population over time. The low rates of second remission (∼40%) and very poor survival after relapse (∼20%) are consistent with this hypothesis.41,42
Genomic studies of large cohorts of diagnostic KMT2A-r infant ALL specimens have revealed a relatively small number of cooperating genomic variants.43-46 A smaller number of paired diagnostic/relapse pairs have been studied, but there appears to be no significant increase or selection of mutations at relapse.44 Thus, it does not appear that relapse in KMT2A-r infant ALL can be explained by the appearance of, or selection for, subclones with resistance-inducing genomic mutations. As discussed in more detail in “Epigenetic agents,” KMT2A-r infant ALL is associated with aberrant epigenetic programs. Emerging data suggest that subclonal epigenetic changes may be responsible for chemoresistance and relapse in KMT2A-r infant ALL,47 raising the possibility that incorporating epigenetically targeted therapies may be able to prevent relapse.
Novel therapies
It is clear that our standard approaches to infant leukemia leave ample room for improvement. The unique molecular biology of KMT2A-r leukemia has suggested novel treatment approaches, several of which are in various stages of clinical investigation. The importance of collaborative and innovative clinical trials for this disease cannot be overemphasized. To that end, the 3 major cooperative groups in Tables 3 and 4 are developing a joint protocol to enhance the ability to test novel treatment approaches and improve the standard of care.
FLT3 inhibitors
KMT2A-r infant ALL is characterized by a distinct global gene-expression profile.48,49 A notable component of this profile is striking overexpression of FLT3.50 FLT3 signaling is constitutively activated in these cases, either by activating mutations51,52 or, more commonly, by autocrine activation via coexpressed FLT3 ligand.53 Moreover, FLT3 tyrosine kinase inhibition results in selective killing of these samples and synergizes with chemotherapy in a sequence-dependent manner.51,53-55 FLT3 overexpression has been shown to confer especially poor prognosis in KMT2A-r infant ALL.56,57 As shown in Table 4, COG trial AALL0631 was the first to incorporate a novel, molecularly targeted agent into frontline treatment of KMT2A-r infant ALL. Unfortunately, the trial failed to demonstrate a benefit for the addition of lestaurtinib, perhaps due in part to pharmacologic limitations.58 Nevertheless, this trial serves as proof of principle that novel targeted therapeutics can feasibly be tested in this high-risk group and has laid the groundwork for the international collaborative trial in development.
Epigenetic agents
Genomic studies have revealed a striking paucity of cooperating genetic alterations in infant KMT2A-r ALL compared with all other subsets of childhood ALL.43-46,59 Accordingly, KMT2A-r infant leukemia is increasingly recognized to be driven by aberrant epigenetic programs. As this complex network of interdependent epigenetic processes is elucidated, novel therapeutic strategies are emerging.
KMT2A translocations include partner genes that recruit multiprotein complexes with chromatin-modifying activity to KMT2A target genes, inducing dysregulated transcription of multiple genes. A required component of this aberrant epigenetic state and KMT2A-r leukemogenesis is the H3K79 methyltransferase DOT1L.60-63 Potent and highly selective small-molecule inhibitors of DOT1L showed promising activity in preclinical models of KMT2A-r leukemia.64,65 Unfortunately, the clinical activity of pinometostat, the first DOT1L inhibitor studied, was limited when used as monotherapy in relapsed adults and children with KMT2A-r leukemia.66,67
BRD4 is one of a number of epigenetic “reader” proteins that binds acetylated histones and facilitates transcription downstream of MYC and other validated oncogenes. A nonbiased RNA interference screen of 243 chromatin-modifying genes identified BRD4 to be required for the maintenance of leukemia in an MLL-AF9 murine model.68 Potent and selective small-molecule inhibitors of BRD4 binding downregulated characteristic KMT2A-r and MYC target genes and demonstrated in vitro and in vivo antileukemic activity by inducing apoptosis and differentiation in murine models, leukemia cell lines, and, most importantly, in a cohort of primary KMT2A-r infant ALL cells.68,69 Early-phase clinical trials in adults with hematologic malignancies have been or are being conducted with bromodomain inhibitors such as OTX015 and CPI-0610. A phase 1 trial of OTX015 in adults with acute leukemia, including 2 with KMT2A-r, demonstrated modest activity as a single agent.70
Also characteristic of KMT2A-r ALL is the epigenetic silencing of another set of genes with tumor-suppressor function via promoter region CpG island hypermethylation.71,72 Increasing degrees of promoter hypermethylation correlated with inferior survival in Interfant-99.72 Demethylating agents such as azacitidine, decitabine, and zebularine preferentially kill KMT2A-r ALL cells, and this correlates with the upregulation of several of the identified silenced genes.71-73 Hypermethylation of certain microRNAs (miRs), such as miR-152, also characterizes infant KMT2A-r ALL and has been associated with inferior outcomes.74,75 Based on these data, as well as the aforementioned report of the critical role of subclonal methylation changes in chemoresistance and relapse of infant KMT2A-r ALL,47 the COG (clinicaltrials.gov NCT02828358) and JPLSG (umin.ac.jp UMIN29275) are each conducting pilot trials of azacitidine in combination with the Interfant chemotherapy backbone.
Deacetylation of histone marks such as H3K9/14 is associated with gene silencing and can be modulated with histone deacetylase (HDAC) inhibitors. Connectivity maps have identified HDAC inhibitors as capable of reversing gene-expression profiles associated with chemotherapy resistance in relapsed childhood ALL76 and in infant KMT2A-r ALL.77 A high-throughput screen of a large panel of US Food and Drug Administration (FDA)-approved and investigational drugs in KMT2A-r infant ALL cell lines identified the HDAC inhibitor romidepsin as efficacious and synergistic in vivo with cytarabine.78 Finally, the HDAC inhibitor panobinostat was found to be effective in xenograft models of KMT2A-r ALL.79 Although demethylating agents and HDAC inhibitors are a promising strategy to reverse the inherent chemoresistance of KMT2A-r infant ALL, a pilot study of decitabine and vorinostat combined with reinduction chemotherapy in children with relapsed ALL led to excessive infectious toxicity,80 so safety has yet to be demonstrated.
Targeting microenvironment interactions
The pattern of remission and early relapse suggests that chemotherapy-resistant leukemia stem cells survive and can recapitulate the leukemia. Interactions between infant KMT2A-r ALL leukemia stem cells and the bone marrow stromal microenvironment via the CXCR4/SDF-1 axis have been shown to mediate survival and therapeutic resistance in KMT2A-r ALL.81 In xenograft models of KMT2A-r infant ALL, CXCR4 inhibition with plerixafor led to markedly enhanced in vivo efficacy of FLT3 inhibitors, suggesting that targeting leukemia-stroma interactions with CXCR4 inhibitors may represent a promising adjunctive therapy. Dynamic upregulation of CXCR4 expression on the surface of acute leukemias (including KMT2A-r cases) in response to cytotoxic chemotherapy may enhance this stroma-mediated resistance and identify patients for whom CXCR4 inhibition may be particularly effective.82 Subsequent preclinical studies have demonstrated the efficacy of small-molecule CXCR4 antagonism in reversing chemoresistance, both in vitro and in vivo,83,84 and these findings led to a clinical trial of plerixafor and chemotherapy in relapsed/refractory pediatric acute leukemia (clinicaltrials.gov NCT01319864). The combination was safe but showed modest clinical activity, although no KMT2A-r ALL patients were enrolled.85
Immunotherapy
The recent FDA approval of blinatumomab86 and tisagenlecleucel87 has generated interest in immunotherapeutic approaches for high-risk B-ALL subsets, to include KMT2A-r infant ALL. A pilot trial of blinatumomab in combination with the Interfant chemotherapy backbone has been initiated by the Interfant group (trialregister.nl NTR6359). One potential limitation, however, is that CD19 antigen is not uniformly expressed in KMT2A-r leukemias. Case reports of immunologic “class switching” from CD19+ lymphoid to CD19− myeloid phenotypes have been reported with CD19-targeted immunotherapies,88-90 and xenograft studies demonstrate leukemia stem/initiating cell activity in CD19− subpopulations in KMT2A-r ALL cases.91-93 Low levels of CD22 expression in KMT2A-r ALL may also limit the activity of inotuzumab.94 Another challenge to chimeric antigen receptor (CAR) T-cell technology in infants is manufacturing product using autologous infant T cells. Recently, a gene-editing technique was used to simultaneously introduce the CD19 CAR construct and disrupt mediators of allogeneic rejection (T-cell receptor α chain and CD52) into healthy donor T cells, creating an “off-the-shelf” universal CAR-T product. Remarkably, 2 relapsed KMT2A-r ALL infants were treated and achieved molecular remission before proceeding to HSCT.95 Ultimately, the success of immunotherapy for infant leukemia will depend upon identifying the right antigen (or group of antigens) to target, because as discussed, CD19 and CD22 may not be optimal.
Targeting RAS pathway
Mutations in NRAS and KRAS have been found in 14% of KMT2A-r infant ALL.96 As has been found in other acute leukemias, RAS mutations tend to be subclonal and not stable between diagnosis and relapse, raising the question of whether they represent leukemia drivers.97,98 However, a cohort in which 70 patients were negative for RAS mutations and 9 were positive demonstrated a worse outcome among the RAS-mutant group,96 and preclinical studies suggest a potential role for MEK inhibition as a targeted therapeutic strategy.99,100
Conclusions and future directions
Infant leukemia is one of most difficult clinical situations encountered in pediatric hematology/oncology. Standard approaches (consensus recommendations summarized in Table 5) are curative in a minority of patients, and participation in clinical trials of novel treatment approaches is strongly encouraged. Although most treatment failures are due to relapse, treatment-related mortality and life-limiting late effects in survivors are also problematic. Recent discoveries regarding the unique biology of these leukemias are fueling the development of a pipeline of exciting novel treatment strategies that are increasingly being incorporated into clinical trials and have the potential to reduce both relapse rates and treatment-related toxicities. Increasing collaboration among the major international cooperative groups will accelerate the translation of biological understanding into better outcomes.
Summary of consensus recommended treatment strategies for infant leukemia subtypes
| Risk group | Defined on the basis of | Recommended treatment approach |
|---|---|---|
| Infant ALL | ||
| High | KMT2A-r, younger age, late MRD clearance | Interfant induction, then intensive chemotherapy consolidation, then strongly consider HSCT (prefer non–total body irradiation based, prefer age at HSCT ≥6 mo); continued consolidation and maintenance if HSCT unavailable |
| Intermediate | KMT2A-r, older age, early MRD clearance | Interfant induction, then intensive chemotherapy consolidation and maintenance |
| Low | wt-KMT2A | Interfant induction, then identical approach as pediatric ALL (risk-stratified chemotherapy based on genetics and MRD response) |
| Infant AML | Identical approach as pediatric AML (intensive chemotherapy/gemtuzumab induction, then risk-based consolidation with chemotherapy/gemtuzumab for low risk and HSCT for high risk) | |
| Risk group | Defined on the basis of | Recommended treatment approach |
|---|---|---|
| Infant ALL | ||
| High | KMT2A-r, younger age, late MRD clearance | Interfant induction, then intensive chemotherapy consolidation, then strongly consider HSCT (prefer non–total body irradiation based, prefer age at HSCT ≥6 mo); continued consolidation and maintenance if HSCT unavailable |
| Intermediate | KMT2A-r, older age, early MRD clearance | Interfant induction, then intensive chemotherapy consolidation and maintenance |
| Low | wt-KMT2A | Interfant induction, then identical approach as pediatric ALL (risk-stratified chemotherapy based on genetics and MRD response) |
| Infant AML | Identical approach as pediatric AML (intensive chemotherapy/gemtuzumab induction, then risk-based consolidation with chemotherapy/gemtuzumab for low risk and HSCT for high risk) | |
Acknowledgment
A.B. was supported by Associazione Italiana per la Ricerca sul Cancro (20564, AIRC 5x1000) and Transcan2-189.
Authorship
Contribution: P.B., R.B., and A.B. wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Patrick Brown, Johns Hopkins Oncology, 1650 Orleans St, CRB1 Room 2M51, Baltimore, MD 21231; e-mail: pbrown2@jhmi.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal