

Key Points
SMN risk was highest in survivors exposed to high-dose unfractionated (600-1200 cGy) or very high-dose fractionated (1440-1750 cGy) TBI.
For low-dose TBI (200-450 cGy), SMN risk was comparable to chemotherapy alone, though still twofold higher than in the general population.

Abstract
We examined the impact of total body irradiation (TBI) dose and fractionation on risk of subsequent malignant neoplasms (SMNs) in the era of reduced-intensity and nonmyeloablative conditioning regimens for hematopoietic cell transplantation (HCT). Among 4905 1-year survivors of allogeneic HCT for hematologic malignancies (N = 4500) or nonmalignant disorders (N = 405) who received transplants between 1969 and 2014, we identified 581 SMNs (excluding squamous and basal cell of skin) in 499 individuals. With a median length of follow-up of 12.5 years, the cumulative incidence of SMNs by 30 years after HCT was 22.0%. Compared with age-, sex-, and calendar year–matched Surveillance, Epidemiology, and End Results (SEER) population rates, the standardized incidence ratio (SIR) of SMNs was increased 2.8-fold. The highest SIRs were for SMNs of bones (SIR, 28.8), oral cavity (SIR, 13.8), skin (SIR, 7.3), central nervous system (SIR, 6.0), and endocrine organs (SIR, 4.9). The highest excess absolute risks (EARs) were seen with breast cancer (EAR, 2.2) and cancers of the oral cavity (EAR, 1.5) and skin (EAR, 1.5) per 1000 person-years. The highest incidence of SMNs was in survivors exposed to unfractionated (600-1000 cGy) or high-dose fractionated (1440-1750 cGy) TBI. For patients receiving low-dose TBI, the incidence was comparable to myeloablative chemotherapy alone, although still twofold higher than in the general population. These data demonstrate a strong effect of TBI dose, dose fractionation, and risk of SMNs after HCT. The cumulative incidence of SMNs increases with follow-up time; thus, HCT survivors require lifetime monitoring for early detection and effective therapy of SMNs.
Introduction
The number of allogeneic hematopoietic cell transplants (HCTs) has increased progressively over the past 2 decades and long-term survival has improved significantly.1,2 Considerable progress has been made in the prevention or attenuation of graft-versus-host disease (GVHD), the most frequent complication after HCT, as well as other conditions that contribute to late mortality3,4 However, with the growing number of patients who are cured of their original disease and survive long-term, the prevalence of posttransplant “subsequent malignant neoplasms” (SMNs) has increased. We and others reported previously on the occurrence of new malignancies after autologous and allogeneic HCT,5-12 documenting significant risks for the development of various malignancies, including, in particular, breast cancer, carcinomas of the oral cavity, tumors of the central nervous system, melanomas, and nonmelanoma skin cancers. Exposure to total body irradiation (TBI) and, for certain cancer types and sites, the presence of chronic GVHD, have been identified as major risk factors. Most patients included in these prior analyses had been conditioned for HCT with high-intensity (“myeloablative”) regimens. However, with the increasing use of low/reduced-intensity (“nonmyeloablative”) regimens over the past 2 decades, the question of to whether these modified regimens would result in a different pattern of long-term complications, including the development of SMNs, has not been addressed. Therefore, we analyzed results in a cohort of 4905 patients conditioned with various intensity regimens in preparation for HCT and surviving for at least 1 year post-HCT in order to examine the differential impact on risk based upon the intensity of different conditioning regimens.
Methods
Patients
Included in the analysis were 4905 patients who underwent allogeneic HCT for malignant or nonmalignant diseases at the Fred Hutchinson Cancer Research Center (Seattle, WA) between 1969 and July 2014 and who had survived at least 1 year after transplantation without developing an SMN. Patients with Fanconi anemia (n = 20) and patients who received transplants for nonhematologic solid tumors (N = 14) were excluded from the analysis. All patients had provided informed consent for follow-up research studies at the time of transplantation.
Conditioning regimen and GVHD prophylaxis
Over the time span of this study, numerous conditioning regimens and GVHD prophylaxis protocols were used. During the earlier study period, most patients received TBI-based regimens with doses of 600 to 1000 cGy, given mostly as a single fraction. Subsequently, doses of 1200 to 1750 cGy were given in multiple fractions, typically in combination with cyclophosphamide (with or without other agents). Until 2001, the radiation source was cobalt; thereafter, radiation has been delivered from a linear accelerator. Some patients received chemotherapy-only conditioning regimens, the majority busulfan-based administered in combination with cyclophosphamide. Beginning in 1997, nonmyeloablative conditioning regimens were used with increasing frequency, consisting of TBI at doses between 200 cGy (single fraction) and 450 cGy (as 1 or 2 fractions) and fludarabine.13
GVHD prophylaxis for patients receiving high-dose (myeloablative) conditioning regimens, similarly, evolved over time as described elsewhere.14,15 For patients receiving nonmyeloablative transplants GVHD prophylaxis included mycophenolate mofetil and a calcineurin inhibitor (cyclosporine or tacrolimus).16 Diagnostic criteria and approaches to therapy for GVHD as well as infection prophylaxis and treatment have been described elsewhere.17,18
Patient follow-up and data collection
Patients are followed for life in the long-term follow-up (LTFU) program under a standardized protocol approved by the institutional review board. Patient and transplant characteristics, conditioning regimen, early post-HCT course, and information on late events, including the development of SMNs, are prospectively collected and maintained in the HCT database. Patients are invited to return to Seattle for a comprehensive post-HCT medical evaluation at 1 year post-HCT and again thereafter, as clinically indicated. In addition, on an annual basis, health status questionnaires are sent to both patients and referring physicians to obtain additional details on a variety of late effects, including SMNs, which are verified by physicians’ reports and, whenever possible, by pathology as well as surgical and other reports for confirmation. As of the cutoff date for this analysis (July 2014) among living survivors, 74% had follow-up contact (patient/physician questionnaires, LTFU clinic) within 2 years and 84% within 3 years. Only 11% had been out of contact for >5 years.
Data on SMNs
Reported malignances were considered to be subsequent to HCT if the histologic classification: (1) was unequivocally different from the diagnosis for which the patient underwent HCT or (2) differed from that of any prior malignancy diagnosed before HCT (for example, a patient with breast cancer who underwent HCT for a secondary acute myeloid leukemia and subsequently developed breast cancer in the same breast after HCT would be considered relapse of pretransplant malignancy and not SMNs). Cases of posttransplant lymphoproliferative disease, typically occurring within the first year after HCT as we reported previously, were excluded from this analysis.19 Squamous cell carcinoma (SCC) of the skin and basal cell carcinoma (BCC) were recorded but will be the subject of a separate report; nonskin SCC are included in this report.
Statistical methods
Patient characteristics, including treatment regimens and diagnoses for which they were undergoing HCT, were summarized using standard descriptive measures.
Patients included had to have survived event-free for 1 year post-HCT; thus, all time-to-event analyses begin at 1 year after HCT. Cumulative incidence estimates to 30 years after HCT were calculated, treating death or second HCT as a competing risk and censoring at last follow-up. Cox proportional hazards modeling was carried out for time to first SMNs using age as the time scale, with time censored at the earliest of age at death, last follow-up, or subsequent HCT. A separate analysis was performed for development of nonskin SCC and similar models were fit for time to first SCC.
Risk factors evaluated in all analyses included sex, race, age at first allogeneic HCT, diagnosis for HCT, stem cell source, preparative regimen (including TBI dose and fractionation), and development of GVHD as a time-dependent covariate. Age at first HCT was categorized into ≤20 years old, >20 to 50 years old, and >50 years old. Primary transplant diagnoses were categorized into 2 groups: nonmalignant conditions and hematologic malignancies. Sources of stem cells for transplant included cord blood, peripheral blood stem cells (PBSCs), and bone marrow (BM). A small number of patients in this analysis received both BM and PBSCs (N = 11) and were included in the PBSC category for analyses. The preparative regimens for HCT were categorized by dose and fractionation of TBI into the following categories: low-dose TBI (ld-TBI; 200-450 cGy given in 1 or 2 fractions), single-fraction TBI (sf-TBI; 600-1000 cGy), fractionated TBI (f-TBI; 600-1200 cGy or 1200-1400 cGy), high-dose f-TBI (1440-1750 cGy), and “chemotherapy only” for those who did not receive TBI. Fourteen 1-year survivors received total marrow irradiation and they were included in the sf-TBI category.
Patients with acute GVHD grades II-IV were categorized as having had “acute GHVD.” Patients were classified with respect to their acute GVHD (only) or chronic GVHD (classic, with or without prior acute GVHD) status as baseline covariates for modeling as of 1 year, and subsequent development of chronic GVHD was modeled as a time-dependent covariate, allowing patients with or without prior acute GVHD to transition to chronic GHVD. Sex, age at first allogeneic HCT, diagnosis for which HCT was performed, source of stem cells, preparative regimen, and acute GVHD status were a priori forced into the models. Race was included in the final model at the α = 0.05 level.
Standardized incidence ratios (SIRs) were calculated as ratios of observed numbers of SMNs to number of expected age-, sex- calendar year–matched US population rates from the Surveillance, Epidemiology, and End Results (SEER) program (SEER Cancer Statistics Review [CSR], 1975-2012 [http://seer.cancer.gov/csr/1975_2012/]). Excess absolute risks (EARs) were calculated as observed minus expected SMNs per 1000 person-years.
Results
Characteristics of 4905 patients eligible for this analysis are shown in Table 1. Median age at time of HCT was 34.5 years (range, 0.3-78.9 years), and median length of follow-up among 1709 patients still living was 12.5 years (range, 1.0-42.11.0 years). Most patients (N = 4500) underwent HCT for a hematologic malignancy (acute and chronic leukemia, lymphoma, and myelodysplastic syndrome) and 405 were transplanted for nonmalignant diseases (severe aplastic anemia, immune deficiencies, hemophagocytic lymphohistiocytosis, hemoglobinopathies, and others). Among the 4905 patients, 499 (11%) developed at least 1 SMN, occurring at a median of 10.3 years (range, 1.0-39.7 years) post-HCT. An additional 9 patients developed an SMN after their second transplant, but were censored at the date of their second transplant. Multiple subsequent malignancies developed in 81 patients (69 had 2, 9 had 3, and 3 had 4). They represented 15 unique sites/histologies, with the most common sites being skin (melanoma, n = 16), breast (n = 13), renal/urinary bladder (n = 8), oral cavity (n = 7), gastrointestinal sites (n = 6), and others (n = 31). Five patients had breast cancer as their first and second SMN, 3 of these occurred in the contralateral breast and the other 2 had new tumors develop 11 and 17 years after the first diagnosis.
Patient characteristics for those who survived and event-free at least 1 year after the first allogeneic transplant
| Risk factor | N (%) |
|---|---|
| Sex | |
| Male | 2814 (57.4) |
| Female | 2091 (42.6) |
| Race | |
| White | 4276 (91.8) |
| Other | 383 (8.2) |
| Age at first allogeneic HCT, y | |
| ≤20 | 1162 (23.7) |
| >20-50 | 2684 (54.7) |
| >50 | 1059 (21.6) |
| HCT diagnosis category | |
| Nonmalignant diseases | 405 (8.3) |
| Hematologic malignancies | 4500 (91.7) |
| Preparative regimen | |
| Chemotherapy only | 1544 (31.6) |
| Low-dose TBI 200-450 cGy | 775 (15.8) |
| Single-fraction TBI 600-1000 cGy | 168 (3.4) |
| Fractionated TBI 600-1200* cGy | 1157 (23.6) |
| Fractionated TBI 1320-1400 cGy | 680 (13.9) |
| Fractionated TBI 1440-1750 cGy | 570 (11.7) |
| Stem cell source | |
| BM | 3184 (64.9) |
| PBSC/BM+PBSC | 1584 (32.3) |
| Cord blood | 137 (2.8) |
| GVHD status before event | |
| None | 856 (18.2) |
| Acute only | 951 (20.2) |
| Chronic only | 752 (16.0) |
| Both acute and chronic | 2157 (45.7) |
| Events after 1 y | |
| In follow-up or subsequent transplant | 3037 (61.9) |
| SMN | 499 (10.2) |
| Death without SMN | 1369 (27.9) |
| Age at first SMN after 1 y, y; categorized, n = 499 | |
| ≤20 | 94 (18.8) |
| >20-50 | 286 (57.3) |
| >50 | 119 (23.9) |
| Year of HCT, 15-y increments | |
| ≤1985 | 759 (15.5) |
| 1986-2000 | 2241 (45.7) |
| ≥2001 | 1905 (38.8) |
| Time between first allogeneic HCT and first SMN, y | |
| ≤10 y since transplant | 123 (24.7) |
| 10.1-20 y since transplant | 189 (37.9) |
| 20.1+ y since transplant | 187 (37.5) |
| Risk factor | N (%) |
|---|---|
| Sex | |
| Male | 2814 (57.4) |
| Female | 2091 (42.6) |
| Race | |
| White | 4276 (91.8) |
| Other | 383 (8.2) |
| Age at first allogeneic HCT, y | |
| ≤20 | 1162 (23.7) |
| >20-50 | 2684 (54.7) |
| >50 | 1059 (21.6) |
| HCT diagnosis category | |
| Nonmalignant diseases | 405 (8.3) |
| Hematologic malignancies | 4500 (91.7) |
| Preparative regimen | |
| Chemotherapy only | 1544 (31.6) |
| Low-dose TBI 200-450 cGy | 775 (15.8) |
| Single-fraction TBI 600-1000 cGy | 168 (3.4) |
| Fractionated TBI 600-1200* cGy | 1157 (23.6) |
| Fractionated TBI 1320-1400 cGy | 680 (13.9) |
| Fractionated TBI 1440-1750 cGy | 570 (11.7) |
| Stem cell source | |
| BM | 3184 (64.9) |
| PBSC/BM+PBSC | 1584 (32.3) |
| Cord blood | 137 (2.8) |
| GVHD status before event | |
| None | 856 (18.2) |
| Acute only | 951 (20.2) |
| Chronic only | 752 (16.0) |
| Both acute and chronic | 2157 (45.7) |
| Events after 1 y | |
| In follow-up or subsequent transplant | 3037 (61.9) |
| SMN | 499 (10.2) |
| Death without SMN | 1369 (27.9) |
| Age at first SMN after 1 y, y; categorized, n = 499 | |
| ≤20 | 94 (18.8) |
| >20-50 | 286 (57.3) |
| >50 | 119 (23.9) |
| Year of HCT, 15-y increments | |
| ≤1985 | 759 (15.5) |
| 1986-2000 | 2241 (45.7) |
| ≥2001 | 1905 (38.8) |
| Time between first allogeneic HCT and first SMN, y | |
| ≤10 y since transplant | 123 (24.7) |
| 10.1-20 y since transplant | 189 (37.9) |
| 20.1+ y since transplant | 187 (37.5) |
98.4% received dose of 1200 cGy.
Cumulative incidence
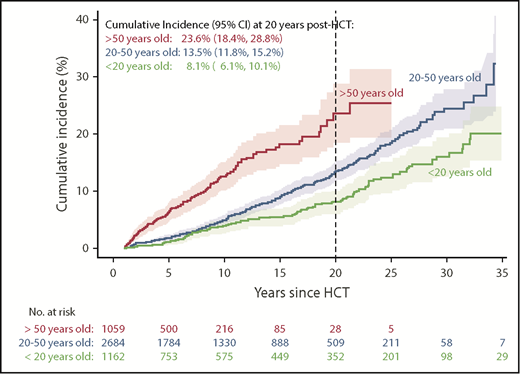
The cumulative incidence of SMNs by 30 years after HCT for the entire cohort was 22.0% (95% confidence interval [CI], 19.8-24.1); the cumulative incidence of SMNs by age at time of HCT and years of follow-up after HCT are shown in Figure 1. At 20 years post-HCT, the cumulative incidence of SMNs for patients <20 years of age at time of HCT was 8.1% (95% CI, 6.1-10.1), 13.5% (95% CI, 11.8-15.2) for those 20 to 50 years, and 23.6% (95% CI, 18.4-28.8) for patients older than 50 years.
SIRs
Risk factors were evaluated via SIRs comparing the number of SMNs observed to those expected, based on person-years and age-, sex-, calendar year–matched US population rates from SEER for different subgroups of survivors, and the EAR per 1000 person-years of follow-up (EAR/1000 PY) was calculated. For the entire cohort, the observed number of SMNs was 2.8-fold higher than expected (95% CI, 2.6, 3.1) and accounted for an EAR of 7.51 cancers per 1000 person-years. The SIR and EAR for all risk factors examined, including sex, age at first allogeneic HCT, transplant diagnosis, preparative regimen, source of stem cells, GVHD status, follow-up time, and current age were significantly elevated (see Table 2). Key findings included the fact that the risk of developing SMNs, even in patients who received chemotherapy-only preparative regimens, was almost twofold higher than in the general population (SIR, 1.9; 95% CI, 1.6, 2.3; EAR, 4.16/1000 PY), and this risk was nearly identical for patients who received ld-TBI (SIR, 2.0; 95% CI, 1.6, 2.6; EAR, 10.63/1000 PY). However, the risk was substantially higher for all other TBI cohorts, including patients exposed to sf-TBI (SIR, 7.8; 95% CI, 5.5, 11.2) and high-dose f-TBI (1440-1750 cGy) (SIR, 5.7; 95% CI, 4.5-7.3), as well as for patients with intermediate-dose radiation exposures, that is, f-TBI at 600 to 1200 and 1320 to 1400 cGy, with SIRs of 3.3 (95% CI, 2.8-3.8) and 3.1 (95% CI, 2.4, 4.0), respectively. Finally, as shown in Figure 2A, the risk of SMNs compared with the general population (SIRs) was higher across every combination of time since transplant and age at time of HCT, but was more influenced by age than by length of follow-up. Survivors who were ≤20 years old at time of HCT had the highest risk of SMNs compared with same age members of the general population, though this risk declined with longer follow-up period relative to HCT. For the older age groups (20-50 years old and >50 years old) at HCT, the SIRs remain elevated to approximately the same degree over time, though follow-up in the >50-years-old cohort is not yet available beyond 20 years. Figure 2B displays the more modest changes in EAR by age at HCT and years since HCT. For survivors <20 years old and those between 2 and 50 years old at time of HCT, the EAR increases with longer length of follow-up.
SIR and EAR (per 1000 person-years) for all SMNs and by risk factors
| Risk factors | Observed | Expected | SIR (95% CI) | EAR (95% CI) |
|---|---|---|---|---|
| All SMNs | 581 | 205 | 2.8 (2.6, 3.1) | 7.5 (6.5, 8.6) |
| Sex | ||||
| Male | 297 | 116.9 | 2.5 (2.2, 2.9) | 6.5 (5.3, 7.9) |
| Female | 284 | 88.1 | 3.2 (2.8, 3.7) | 8.8 (7.3, 10.6) |
| Age at first allogeneic HCT, y | ||||
| ≤20 y | 110 | 8.8 | 12.5 (10.2, 15.3) | 10.6 (5.9, 16.7) |
| >20-50 y | 327 | 112.1 | 2.9 (2.6, 3.3) | 13.4 (8.8, 20.1) |
| >50 y | 144 | 84.1 | 1.7 (1.4, 2.0) | 8.6 (6.7, 10.8) |
| HCT diagnosis category | ||||
| Nonmalignant disorder | 34 | 13.2 | 2.6 (1.8, 3.7) | 8.0 (5.2, 11.5) |
| Hematologic/lymphoid malignancy | 547 | 191.8 | 2.9 (2.6, 3.1) | 8.7 (6.3, 11.7) |
| Preparative regimen | ||||
| Chemotherapy only | 144 | 75.9 | 1.9 (1.6, 2.3) | 6.9 (5.9, 8.03) |
| Low-dose TBI 200-450 cGy | 63 | 31.1 | 2.0 (1.6, 2.6) | 4.2 (2.7, 5.9) |
| Single-dose TBI 600-1000 cGy | 37 | 4.7 | 7.8 (5.5, 11.2) | 8.6 (6.8, 10.7) |
| Fractionated TBI 600-1200 cGy | 183 | 56.4 | 3.3 (2.8, 3.8) | 7.6 (5.5, 10.1) |
| Fractionated TBI 1320-1400 cGy | 68 | 22 | 3.1 (2.4, 4.0) | 7.6 (5.1, 10.5) |
| Fractionated TBI 1440-1750 cGy | 81 | 14.2 | 5.7 (4.5, 7.3) | 4.9 (3.2, 6.8) |
| Stem cell source | ||||
| BM | 431 | 143.6 | 3.0 (2.7, 3.3) | 10.6 (7.7, 14.1) |
| PBSC/BM+PBSC | 144 | 60.7 | 2.4 (2.0, 2.8) | 11.6 (4.1, 29.1) |
| Cord | 6 | 0.7 | 8.7 (3.7, 20.4) | 3.2 (1.6, 55) |
| GVHD status* | ||||
| None | 91 | 34.7 | 2.6 (2.1, 3.3) | 5.1 (3.4, 7.2) |
| Acute only | 100 | 32.4 | 31 (25, 3.8) | 7.6 (5.4, 10.2) |
| Chronic (± acute) | 353 | 131.6 | 3.0 (2.4, 3.0) | 8.2 (6.8, 9.7) |
| Follow-up time after first allogeneic HCT,*y | ||||
| ≤10 | 396 | 142.1 | 2.8 (2.5, 3.1) | 6.8 (5.7, 8.0) |
| >10-20 | 119 | 44.3 | 2.7 (2.2, 3.3) | 8.0 (5.7, 10.7) |
| >20-30 | 59 | 16.5 | 3.6 (2.7, 4.7) | 13.5 (9.0, 19.44) |
| >30 | 7 | 2 | 3.4 (1.7, 7.2) | 13.7 (3.6, 34.7) |
| Current age,*y | ||||
| ≤20 | 37 | 1.2 | 30.9 (22.6, 42.4) | 5.4 (3.9, 7.5) |
| >20-50 | 239 | 53 | 4.5 (3.9, 5.2) | 6.4 (5.3, 7.5) |
| >50 | 305 | 150.8 | 2.0 (1.8, 2.3) | 10.9 (8.5, 13.7) |
| Risk factors | Observed | Expected | SIR (95% CI) | EAR (95% CI) |
|---|---|---|---|---|
| All SMNs | 581 | 205 | 2.8 (2.6, 3.1) | 7.5 (6.5, 8.6) |
| Sex | ||||
| Male | 297 | 116.9 | 2.5 (2.2, 2.9) | 6.5 (5.3, 7.9) |
| Female | 284 | 88.1 | 3.2 (2.8, 3.7) | 8.8 (7.3, 10.6) |
| Age at first allogeneic HCT, y | ||||
| ≤20 y | 110 | 8.8 | 12.5 (10.2, 15.3) | 10.6 (5.9, 16.7) |
| >20-50 y | 327 | 112.1 | 2.9 (2.6, 3.3) | 13.4 (8.8, 20.1) |
| >50 y | 144 | 84.1 | 1.7 (1.4, 2.0) | 8.6 (6.7, 10.8) |
| HCT diagnosis category | ||||
| Nonmalignant disorder | 34 | 13.2 | 2.6 (1.8, 3.7) | 8.0 (5.2, 11.5) |
| Hematologic/lymphoid malignancy | 547 | 191.8 | 2.9 (2.6, 3.1) | 8.7 (6.3, 11.7) |
| Preparative regimen | ||||
| Chemotherapy only | 144 | 75.9 | 1.9 (1.6, 2.3) | 6.9 (5.9, 8.03) |
| Low-dose TBI 200-450 cGy | 63 | 31.1 | 2.0 (1.6, 2.6) | 4.2 (2.7, 5.9) |
| Single-dose TBI 600-1000 cGy | 37 | 4.7 | 7.8 (5.5, 11.2) | 8.6 (6.8, 10.7) |
| Fractionated TBI 600-1200 cGy | 183 | 56.4 | 3.3 (2.8, 3.8) | 7.6 (5.5, 10.1) |
| Fractionated TBI 1320-1400 cGy | 68 | 22 | 3.1 (2.4, 4.0) | 7.6 (5.1, 10.5) |
| Fractionated TBI 1440-1750 cGy | 81 | 14.2 | 5.7 (4.5, 7.3) | 4.9 (3.2, 6.8) |
| Stem cell source | ||||
| BM | 431 | 143.6 | 3.0 (2.7, 3.3) | 10.6 (7.7, 14.1) |
| PBSC/BM+PBSC | 144 | 60.7 | 2.4 (2.0, 2.8) | 11.6 (4.1, 29.1) |
| Cord | 6 | 0.7 | 8.7 (3.7, 20.4) | 3.2 (1.6, 55) |
| GVHD status* | ||||
| None | 91 | 34.7 | 2.6 (2.1, 3.3) | 5.1 (3.4, 7.2) |
| Acute only | 100 | 32.4 | 31 (25, 3.8) | 7.6 (5.4, 10.2) |
| Chronic (± acute) | 353 | 131.6 | 3.0 (2.4, 3.0) | 8.2 (6.8, 9.7) |
| Follow-up time after first allogeneic HCT,*y | ||||
| ≤10 | 396 | 142.1 | 2.8 (2.5, 3.1) | 6.8 (5.7, 8.0) |
| >10-20 | 119 | 44.3 | 2.7 (2.2, 3.3) | 8.0 (5.7, 10.7) |
| >20-30 | 59 | 16.5 | 3.6 (2.7, 4.7) | 13.5 (9.0, 19.44) |
| >30 | 7 | 2 | 3.4 (1.7, 7.2) | 13.7 (3.6, 34.7) |
| Current age,*y | ||||
| ≤20 | 37 | 1.2 | 30.9 (22.6, 42.4) | 5.4 (3.9, 7.5) |
| >20-50 | 239 | 53 | 4.5 (3.9, 5.2) | 6.4 (5.3, 7.5) |
| >50 | 305 | 150.8 | 2.0 (1.8, 2.3) | 10.9 (8.5, 13.7) |
Time-dependent factors.
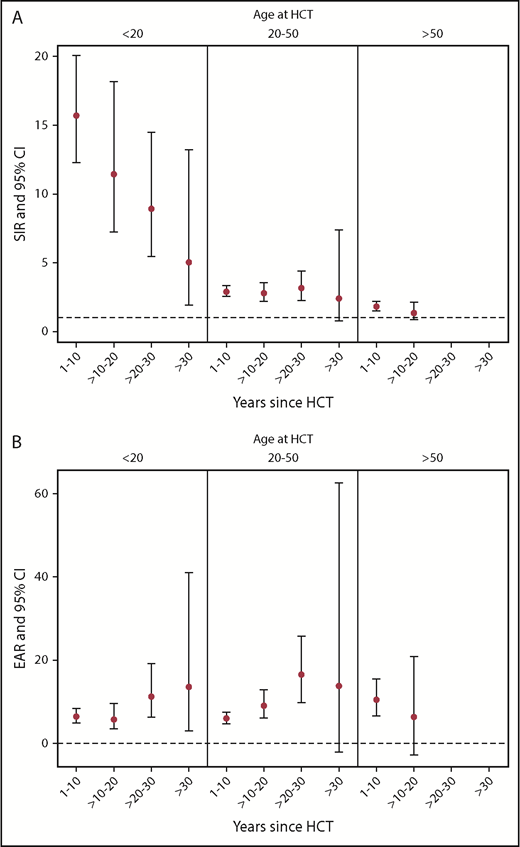
SIRs and EARs by years since HCT and age at time of transplant for development of SMNs. (A) SIRs and 95% CIs. (B) EAR per 1000 person-years and 95% CIs.
SIRs and EARs by years since HCT and age at time of transplant for development of SMNs. (A) SIRs and 95% CIs. (B) EAR per 1000 person-years and 95% CIs.
Figure 3 summarize the cancer site–specific SIRs and EARs (further diagnostic subcategories are detailed in supplemental Table 1, available on the Blood Web site). For the majority of cancer sites, the observed number of cases was significantly higher than expected compared with the general population, with the highest SIRs seen for cancers of the bones/joints, oral cavity/pharynx, skin (majority melanoma), brain, and endocrine glands (majority thyroid). The highest EARs were seen with breast cancer (EAR, 2.2; 95% CI, 1.4, 3.0) and cancers of the oral cavity (EAR, 1.5; 95% CI, 1.2, 2.0) and skin (EAR, 1.5; 95% CI, 1.5, 2.0) per 1000 person-years of follow-up.
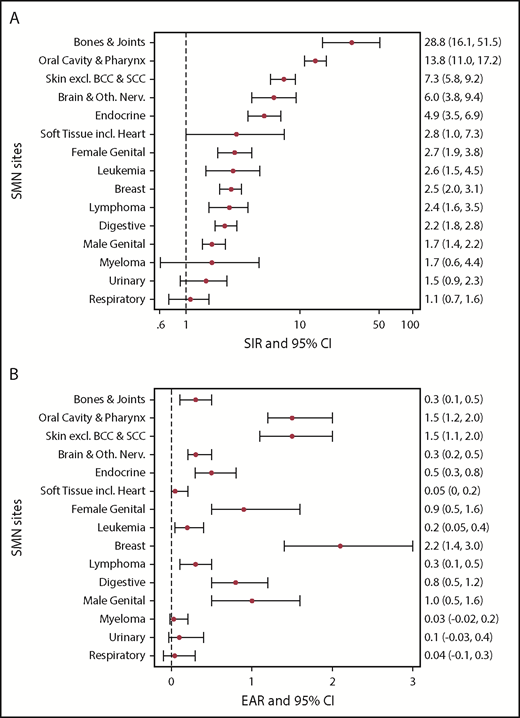
Site-specific SIRs and EAR for development of SMNs. (A) SIRs and 95% CIs. (B) EAR per 1000 person-years and 95% CIs. excl., excluding; incl., including; Oth. Nerv., other nervous system.
Site-specific SIRs and EAR for development of SMNs. (A) SIRs and 95% CIs. (B) EAR per 1000 person-years and 95% CIs. excl., excluding; incl., including; Oth. Nerv., other nervous system.
Risk factors for development of subsequent neoplasms
Results of the Cox proportional hazards model for all SMNs are shown in Table 3. HCT recipients of white race were more likely to develop SMNs compared with other races (hazard ratio [HR], 1.75; 95% CI, 1.04-2.94; P = .04). Younger age was a significant risk factor. Patients who were ≤20 years old at the time of HCT had a 2.28-fold higher risk of SMNs (95% CI, 1.31-3.96; P = .003) than those who were older than 50 years at time of HCT. The stem cell source also impacted the risk of SMNs. Recipients of cord blood (HR, 3.02; 95% CI, 1.29-7.09; P = .01) or PBSC (HR, 1.55; 95% CI, 1.16-2.07; P, .003) had a higher risk of SMNs than patients receiving BM, even after adjustment for acute and chronic GVHD status. The development of acute GVHD was associated with a higher overall risk of SMNs (HR, 1.37; 95% CI, 1.00-1.88; P = .05). Patient sex and diagnosis (malignant vs nonmalignant) did not prove to be significant risk factors.
Cox proportional hazards model for all SMNs, for patients who were alive 1 year posttransplant
| Risk factor | HR | 95% lower CI | P |
|---|---|---|---|
| Race | |||
| Other | 1.0 | Ref | Ref |
| White | 1.8 | (1.0, 2.9) | .04 |
| Sex | |||
| Female | 1.0 | Ref | Ref |
| Male | 0.9 | (0.7, 1.1) | .24 |
| Age at first allogeneic HCT | |||
| >50 y | 1.0 | Ref | Ref |
| >20-50 y | 1.2 | (0.97-1.8) | .24 |
| ≤20 y | 2.3 | (1.3, 4.0) | .003 |
| HCT diagnosis category | |||
| Nonmalignant diseases | 1.0 | Ref | Ref |
| Hematologic malignancies | 1.0 | (0.7, 1.6) | .90 |
| Stem cell source | |||
| BM | 1.0 | Ref | Ref |
| Cord | 3.0 | (1.3, 7.1) | .01 |
| PBSC ± BM | 1.6 | (1.2, 2.1) | .003 |
| Preparative regimen | |||
| Chemotherapy only | 1.0 | Ref | Ref |
| Low-dose TBI 200-450 cGy | 1.2 | (0.8, 1.7) | .42 |
| Single-fraction TBI 600-1000 cGy | 3.2 | (1.9, 5.3) | <.0001 |
| Fractionated TBI 600-1200 cGy | 1.7 | (1.3, 2.2) | .0004 |
| Fractionated TBI 1320-1400 cGy | 1.6 | (1.1, 2.2) | .02 |
| Fractionated TBI 1440-1750 cGy | 2.1 | (1.5, 3.1) | <.0001 |
| GVHD status | |||
| None | 1.0 | Ref | Ref |
| Acute only | 1.4 | (1.0, 1.9) | .05 |
| Chronic ± acute | 1.2 | (1.0, 1.6) | .10 |
| Risk factor | HR | 95% lower CI | P |
|---|---|---|---|
| Race | |||
| Other | 1.0 | Ref | Ref |
| White | 1.8 | (1.0, 2.9) | .04 |
| Sex | |||
| Female | 1.0 | Ref | Ref |
| Male | 0.9 | (0.7, 1.1) | .24 |
| Age at first allogeneic HCT | |||
| >50 y | 1.0 | Ref | Ref |
| >20-50 y | 1.2 | (0.97-1.8) | .24 |
| ≤20 y | 2.3 | (1.3, 4.0) | .003 |
| HCT diagnosis category | |||
| Nonmalignant diseases | 1.0 | Ref | Ref |
| Hematologic malignancies | 1.0 | (0.7, 1.6) | .90 |
| Stem cell source | |||
| BM | 1.0 | Ref | Ref |
| Cord | 3.0 | (1.3, 7.1) | .01 |
| PBSC ± BM | 1.6 | (1.2, 2.1) | .003 |
| Preparative regimen | |||
| Chemotherapy only | 1.0 | Ref | Ref |
| Low-dose TBI 200-450 cGy | 1.2 | (0.8, 1.7) | .42 |
| Single-fraction TBI 600-1000 cGy | 3.2 | (1.9, 5.3) | <.0001 |
| Fractionated TBI 600-1200 cGy | 1.7 | (1.3, 2.2) | .0004 |
| Fractionated TBI 1320-1400 cGy | 1.6 | (1.1, 2.2) | .02 |
| Fractionated TBI 1440-1750 cGy | 2.1 | (1.5, 3.1) | <.0001 |
| GVHD status | |||
| None | 1.0 | Ref | Ref |
| Acute only | 1.4 | (1.0, 1.9) | .05 |
| Chronic ± acute | 1.2 | (1.0, 1.6) | .10 |
HR, hazard ratio; ref, reference.
Radiation dose and fractionation
To further explore the impact of radiation, TBI doses were grouped according to dose and fractionation schedule and results were compared with those in patients receiving chemotherapy-only conditioning (primarily busulfan-based regimens) (Table 3). The risk of SMNs was highest in patients who received sf-TBI 600 to 1000 cGy (HR, 3.18; 95% CI, 1.92-5.26; P < .0001). Patients who received the highest cumulative dose of f-TBI (1440-1750 cGy) had the next highest risk of SMNs with an HR of 2.14 (95% CI, 1.49-3.06; P < .0001). In the 2 intermediate-dose categories (f-TBI, 600-1200 cGy, and f-TBI, 1320-1400 cGy), the risks were somewhat attenuated but still significantly increased with HRs of 1.67 (95% CI, 1.26-2.21; P = .0004) and 1.55 (95% CI, 1.09-2.21; P = .02), respectively, compared with patients given chemotherapy only. In contrast, in recipients who received TBI doses of 200 to 450 cGy, the risk of SMNs was not significantly different from that in patients who received chemotherapy-only preparative regimens (HR, 1.17; 95% CI, 0.8-1.72; P = .42).
SCC
In this cohort, 89 patients developed a nonskin SCC (any site) >1 year after HCT. In a multivariable Cox regression analysis for the risk of developing SCC in nonskin sites, chronic GHVD (with or without preceding acute) was the strongest risk factor, with a 4.9-fold (95% CI, 1.9-12.2; P = .001) increase in risk compared with patients who did not develop GVHD. Another significant risk factor was male sex (HR, 1.6; 95% CI, 1.0-2.5; P = .045). The risk declined with increasing age at HCT. Notably, there was no association with TBI at any dose and an increased risk of nonskin SCC.
Discussion
The primary aim of this study was to determine the impact of the intensity and quality of the conditioning regimen on the risk of developing SMNs after allogeneic HCT. The analysis focused on the effect of ld-TBI as used in patients who received transplants after nonmyeloablative conditioning, in comparison with patients who received high-dose TBI (sf-TBI or higher doses of f-TBI) regimens or those who were conditioned with chemotherapy only. Overall results confirmed data from earlier reports, showing an incidence of malignancies 2.8-fold higher than expected. The most significant risk factor was high-dose TBI. In contrast, the use of ld-TBI (200-450 cGy) was associated with a risk comparable to that observed with chemotherapy-only regimens, which was approximately twofold higher than in the general population. Significant risk factors in addition to TBI were white race, younger age at HCT, and the use of cord blood or PBSCs rather than BM. Although not significant for all SMNs, the development of chronic GVHD (with or without prior acute GVHD) was the most important risk factor for nonskin SCC.
The effects of TBI dose and dose fractionation on risk of SMNs were striking. Similar to previous data,5,20,21 TBI given as a single exposure (dose range, 600-1000 cGy) carried the highest risk for SMNs, nearly eightfold higher than seen in the general population. Administration of TBI in smaller fractions (120-200 cGy) reduced the risk of SMNs at lower cumulative doses, but any benefit of fractionation was lost with cumulative doses ≥1440 cGy. As shown by Withers22 and others, dose fractionation, in a time-dependent fashion, allows for interfraction DNA repair, albeit incomplete, in many target tissues. It is likely, however, that with progressively higher total doses, the residual damage accumulates to a level similar to that seen with single-dose TBI. Although the risk of SMNs was significantly reduced at very low TBI doses (200-450 cGy), it was comparable to the risk associated with chemotherapy-only conditioning, which carries a risk approximately twofold higher than in the general population. This observation is consistent with the concept that any cytotoxic therapy, be it chemotherapy or ionizing radiation, is associated with tissue injury that predisposes to the development of malignancies. In addition to direct cellular damage, radiation profoundly alters gene-expression levels, which in turn leads to changes in proinflammatory cytokines and changes in the redox milieu, setting the stage for disturbed tissue repair and proliferation.23,24,25
Another significant risk factor was patient age. Younger age (≤20 years) at the time of HCT was associated with a more than twofold higher risk of SMNs than in patients who received transplants as older adults (>50 years of age), and was 30-fold higher than expected in the age- and sex-matched general population. Although progressively older age was associated with lower SIRs compared with younger ages, there was no apparent attenuation of that risk over time post-HCT. Although the time interval elapsed because HCT did not have as significant an impact as patient age at time of HCT, the present data underscore the need for ongoing long-term monitoring of HCT survivors and the need for patient education regarding their long-term risks.
Also of note was the fact that the overall risk of SMNs was higher among patients of white race than among other racial groups. We had previously observed a higher incidence of BCCs among patients of lighter skin races.9 However, the present analysis excluded nonmelanoma skin cancers and it is not immediately apparent why the incidence for all SMNs should be higher in white patients. Conceivably, this finding was related to race-associated polymorphisms, for example, in DNA repair genes that rendered white patients more sensitive to the cytotoxic insult from the preparative regimens or that interfered with tissue repair.26
Another risk factor was the use of PBSCs or cord blood cells (adjusted for acute/chronic GVHD). Although there were only 6 SMNs in cord blood recipients (4 solid tumors, 2 non-Hodgkin lymphomas), too few for a detailed analysis, there were 124 SMNs in recipients of PBSC grafts. SMNs occurred in 32 different sites, although the 3 most common malignancies were melanoma (24%), carcinoma of the prostate (15%), and breast cancer (9%). The reason for this is not immediately apparent.
Previous reports on patients undergoing autologous transplantation for non-Hodgkin lymphoma had noted a higher incidence of post-HCT myelodysplastic syndrome or acute myeloid leukemia with the use of mobilized PBSCs than with BM cells.27 In that setting, the result was thought to be related to a high concentration of hematopoietic precursor cells previously exposed to cytotoxic chemotherapy. Such an explanation would not be valid with allogeneic HCT, from which hematopoietic stem cells are derived from healthy donors, and, in fact, hematologic malignancies were rare. However, PBSCs have been shown to exert a more potent allogeneic effect and lead to the development of chronic GVHD more frequently than cells obtained directly from the marrow.28 Furthermore, post-HCT immunosuppression for GVHD tends to be required longer than in patients given marrow as a source of stem cells.29 Although in the present study chronic GVHD was a significant factor only for the development of nonskin SCCs (HR, 4.9; P = .001), the absence of clinically apparent GVHD does not preclude the presence of an effect of subclinical GVHD, which has been described in earlier studies. Those studies also suggested that the amount of treatment given for chronic GVHD was associated with an increased risk of SMNs, in particular, SCC of the oral cavity.30
The broad spectrum of different sites and histologies of SMNs in this study was similar to that described in previous reports.6 These findings have significant implications from a cancer-screening standpoint, as the wide spectrum of malignancies prohibits the development of any uniform screening recommendations. At a minimum, adult survivors after HCT should undergo all standardized cancer-screening recommendations that exist for the general population for breast, colorectal, cervical, and lung cancer. In addition, annual examinations of skin and oral cavity are strongly encouraged. For pediatric survivors, recommendations are even more challenging, but LTFU guidelines developed by the Children’s Oncology Group (http://www.survivorshipguidelines.org/) based on treatment exposures are available and provide guidance for those caring for these young survivors.
There are several shortcomings to the present study, as for many, if not all retrospective analyses. Although the follow-up of our patients is rather complete, there is still the possibility of ascertainment bias as we cannot be certain that absolutely all SMNs were reported. Additionally, the follow-up of patients exposed to ld-TBI regimens remains shorter than for patients given high-dose TBI, although we now have over 20 years of follow-up even for that subgroup. All cases of hematopoietic SMNs were reviewed by the author (K.S.B.) in order to ascertain relapse vs new unique malignancy (in patients who had their original transplant for a hematopoietic malignancy). When available, donor vs host chimerism of the hematopoietic malignancy or cytogenetics were used in making the determination. Also, the cutoffs used for TBI dose ranges were somewhat arbitrary, but these categories were based on doses used in various TBI-based protocols over time. Nevertheless, the wide dose range examined offers assurance that there was, indeed, a clear dose dependence for tumor induction. Given that there was a relatively small number of children who received the ld-TBI regimens, we cannot confidently assume that their risks over time will be the same as seen in adults. Finally, because tumor tissue suitable for molecular analysis was not available in the majority of cases, it was not possible to establish a relationship between a particular TBI dose and the type of cellular injury, including induction of mutations. Data were not available to assess other risk factors such as pre- or post-HCT UV light exposure, or tobacco and alcohol use, which are known potential risk factors, particularly for the development of skin and head and neck cancers, respectively. Other pre-HCT treatment exposures may have contributed to the overall risk but information was not available for this analysis. It is known that cancer survivors who have not undergone HCT are at an increased risk of developing SMNs, and at least 1 study comparing conventionally treated childhood cancer survivors to survivors who received HCT found greater than an eightfold higher risk of SMNs in those who had undergone HCT.31 Future studies should account for treatment exposures prior to HCT.
In summary, the present results confirm the elevated risk of developing SMNs for all HCT survivors, with TBI exposure being a major risk factor. However, the impact of TBI was strongly dose- and fractionation-dependent, and TBI doses used for nonmyeloablative conditioning regimens did not increase the risk beyond that observed with myeloablative chemotherapy-only–based regimens. Given the effectiveness of the ld-TBI in promoting engraftment and reducing relapse risk without significant added toxicity when combined with chemotherapy-based regimens,32 there does not appear to be a rationale to try and avoid its use based on any increased risk of SMNs, a finding of particular importance for children who require HCT. Other factors not accounted for in this analysis including pretransplant therapies, genetic predisposition, and family history, lifestyle, and immune competence are also likely important factors and should be evaluated in future studies.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This work was supported by the National Cancer Institute under award numbers P01CA018029 and P01CA078902 and by the National Heart, Lung, and Blood Institute under award number P01HL122173 from the National Institutes of Health (Bethesda, MD).
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health, which had no involvement in the study design, the collection, analysis, and interpretation of data, the writing of the report, nor in the decision to submit the article for publication.
Authorship
Contribution: K.S.B., W.M.L., B.M.S., and H.J.D. designed, directed, and performed research, analyzed data, and wrote the manuscript; G.S. provided data sets for analysis; W.M.L. and P.J.G. performed statistical analysis and wrote the manuscript; R.P.E., M.E.F., and R.S. reviewed data and the manuscript; and all authors critically reviewed and approved the manuscript prior to submission.
Conflict-of interest disclosure: The authors declare no competing financial interests.
Correspondence: K. Scott Baker, Fred Hutchinson Cancer Research Center, 1100 Fairview Ave N, Seattle, WA 98109; e-mail: ksbaker@fredhutch.org.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal