

Key Points
An endogenous, dysfunctional (CRM+) FIX molecule affects prophylactic FIX efficacy.
Recovery studies indicate the amount of extravascular, Col4-bound FIX is several fold greater than the FIX in plasma.

Abstract
Factor IX (FIX) binds to collagen IV (Col4) in the subendothelial basement membrane. In hemophilia B, this FIX-Col4 interaction reduces the plasma recovery of infused FIX and plays a role in hemostasis. Studies examining the recovery of infused BeneFix (FIXWT) in null (cross-reactive material negative, CRM−) hemophilia B mice suggest the concentration of Col4 readily available for binding FIX is ∼405 nM with a 95% confidence interval of 374 to 436 nM. Thus, the vascular cache of FIX bound to Col4 is several-fold the FIX level measured in plasma. In a mouse model of prophylactic therapy (testing hemostasis by saphenous vein bleeding 7 days after infusion of 150 IU/kg FIX), FIXWT and the increased half-life FIXs Alprolix (FIXFC) and Idelvion (FIXAlb) produce comparable hemostatic results in CRM− mice. In bleeding CRM− hemophilia B mice, the times to first clot at a saphenous vein injury site after the infusions of the FIX agents are significantly different, at FIXWT < FIXFC < FIXAlb. Dysfunctional forms of FIX, however, circulate in the majority of patients with hemophilia B (CRM+). In the mouse prophylactic therapy model, none of the FIX products improves hemostasis in CRM+ mice expressing a dysfunctional FIX, FIXR333Q, that nevertheless competes with infused FIX for Col4 binding and potentially other processes involving FIX. The results in this mouse model of CRM+ hemophilia B demonstrate that the endogenous expression of a dysfunctional FIX can deleteriously affect the hemostatic response to prophylactic therapy.
Introduction
Individuals with hemophilia B are deficient in coagulation factor IX (FIX). In the body, FIX distributes between plasma and another readily accessible extravascular location. This extravascular binding initially attributed to an unknown molecule on the surface of the endothelium,1,2 was subsequently shown to be type IV collagen (Col4).3 Col4 is the only collagen restricted exclusively to, and a prominent component of, the basement membrane of all cells.4 The equilibration between FIX in plasma and FIX bound to vascular Col4 is fast and reversible.5,6 Therefore, the recovery in plasma of FIX infused into a patient with hemophilia B is only 40% to 50% of that anticipated in the absence of the vascular Col4 binding sites.7
We demonstrated that the γ-carboxyglutamic acid (Gla) domain of FIX is solely responsible for its interaction with Col4,8 and that lysine at position 5 and valine at position 10 within the Gla domain are crucial for this interaction. For example, mutations at lysine 5 (FIXK5A) or valine 10 (FIXV10K) substantially reduce the affinity of FIX for Col4, whereas FIXK5R binds Col4 with 3- to 5-fold increased affinity.9 Remarkably, all these tested mutant FIX molecules have normal or increased activity in in vitro phospholipid-dependent coagulation assays. That the extravascular binding of FIX to Col4 plays a functional role in hemostasis is demonstrated by 3 observations: postinfusion recovery in mice of FIXK5A > > FIXWT > FIXK5R,10 bleeding in FIX null mice after a saphenous vein injury is significantly less 7 days after infusion of FIXK5R than after infusion of FIXK5A (when plasma FIX levels are undetectable),11 and knock-in mice expressing FIXK5A have a clear, modest bleeding diathesis.12,13
There is considerable interpatient variability in the recovery of infused FIX in patients with hemophilia B, leading to the recommendation that FIX recovery be empirically determined in individual patients. Missense mutations in the FIX gene occur in 75% of patients with hemophilia B and in 60% of those with severe hemophilia B.14,15 These missense mutations frequently associate with the production of dysfunctional FIX proteins that are detectable by immunologic antigen assays as cross-reactive material (CRM+). Therefore, most patients with hemophilia B are CRM+, rather than null for FIX expression (CRM−). There is evidence for enhanced plasma recovery of FIX in some CRM+ patients compared with CRM− patients.16-18 Increased recovery is expected if the endogenous, dysfunctional FIX occupies Col4 binding sites, thereby competing with the infused FIX for binding to Col4.
Here, we compare the hemostatic effects of a variety of FIX products in mouse models of FIX CRM− (null), and CRM+ hemophilia B. Empiric results for the recovery of human FIX in hemophilia mice were used to estimate the quantity of readily accessible vascular Col4 capable of binding FIX.
Methods
Mice
Mouse experiments were approved by the Institutional Animal Care and Use Committee at the University of North Carolina at Chapel Hill and followed Public Health Service guidelines for animal care and use. FIX null (CRM−)19 and hFIXR333Q knock-in6 mice (CRM+) with hemophilia B and normal control mice from Jackson Labs were all in the C57BL/6 genetic background.
Proteins
The Pfizer Corporation provided BeneFix (FIXWT). Alprolix (FIXFC) and Idelvion (FIXAlb) were from Diversified Biologicals (Miami, FL). The proteins were reconstituted per the manufacturer’s instructions immediately before infusion, and a new vial was used for each experiment.20 FXIa was from Haematologic Technologies, Essex Junction, VT.
Human FIX ELISA
FIX was quantified by sandwich enzyme-linked immunosorbent assay (ELISA) performed at Green Mountain Antibodies (Burlington, VT). Ninety-six-well plates were coated overnight at 4°C with 100 µL per well of a mouse monoclonal antibody to the heavy chain of human FIX (GMA-102) at 2 μg/mL in 0.2 M in carbonate-bicarbonate buffer at pH 9.4 (Pierce #28382). Coated plates were blocked for 1 hour at 4°C with blocking buffer (0.1% bovine serum albumin, 20 mM sodium phosphate, 0.15 M sodium chloride, 0.05% Tween20), 300 µL per well. Plates were washed 3 times in PBST (20 mM sodium phosphate at pH 7.4, 0.15 M sodium chloride, 0.05% Tween20), and serial dilutions of mouse plasma in blocking buffer (100 µL per well) were applied to blocked plates for 1 hour at room temperature. After washing as described earlier, plates were incubated for 1 hour at room temperature with 100 µL per well biotinylated mouse monoclonal antibody to the light chain of FIX (GMA-184-BIO) at 1 μg/mL in blocking buffer. Plates were washed as described earlier and incubated for 30 minutes at room temperature with 100 µL per well streptavidin HRP (Jackson ImmunoResearch, catalog #016-030-084) diluted 1:5000 in blocking buffer. After a final washing step, plates were developed with 100 µL per well o-phenylenediamine dihydrochloride in the dark for 5 minutes, stopped with 50 µL per well 2 M H2SO4, and read at 490 nm on a microplate reader. FIX concentrations in mouse plasma samples were determined by comparison with a standard curve produced with FIX (Enzyme Research Laboratories, HFIX 1009) in human FIX-deficient plasma (George King Biomedical 0900). All samples were tested at least twice, and the mean values were reported.
Estimation of total Col4 available for binding human FIXWT in FIX null mice
Blood samples were obtained from the saphenous vein 10 minutes after intravenous infusion of variable quantities of FIXWT. Our calculations assume that the FIX remaining in the plasma is that not bound to Col4, and that the FIX lost during the initial 10 minutes has formed a complex with Col4. In our model, concentrations of infused FIX, plasma FIX, and Col4-bound FIX are calculated as moles per plasma volume.21 The data were fit to FIXt-C by the quadratic equation for chemical equilibrium:

where C represents the calculated complex between FIX and Col4, FIXt is total FIX, Col4t is total Col4 available to bind FIX, Kd is the equilibrium dissociation constant for the FIX-Col4 interaction, and the plasma concentration is FIXt-C (FIXt is calculated as though all infused FIX remained in the plasma volume). The least squares fit was obtained using Mathematica version 11.3, by several different methods, Quasi-Newton, PrincipalAxis, Levenburg-Marquardt, and GradientAxis; all yielded the same results. The error bars in Figure 1 represent the standard deviations of the mean calculated individually from all the data points for each infused FIX concentration (n = at least 8). The confidence interval was calculated as described.22
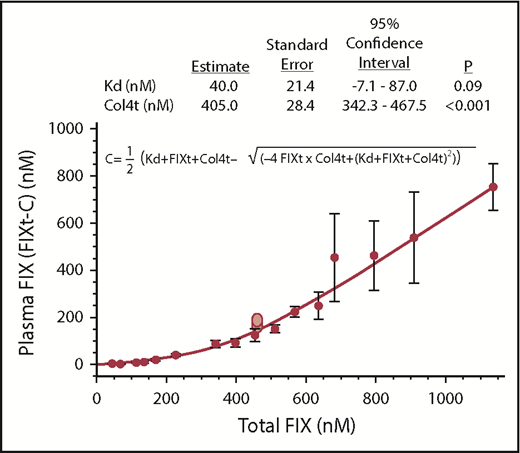
Estimation of vascular Col4 available for binding FIX. Blood samples were taken 10 minutes after infusion of different doses (8, or more, mice per dose) of FIXWT into CRM− mice. The data were fit to FIXt-C (the quadratic equation for equilibrium) and were plotted as the total infused FIX (x-axis) vs measured plasma FIX (y-axis). The empirically determined FIX concentrations were determined by ELISA (Methods).The plotted line is the fit of the experimental data (FIXt-C), where C is the calculated complex. The error bars represent the standard deviation of the mean for 8 or more mice for each level of infused FIX. The 2 light red points in the figure represent data collected in our laboratory about 5 years ago, and is only included as in internal check. The table above the figure represents the statistics of the quadratic fit.
Estimation of vascular Col4 available for binding FIX. Blood samples were taken 10 minutes after infusion of different doses (8, or more, mice per dose) of FIXWT into CRM− mice. The data were fit to FIXt-C (the quadratic equation for equilibrium) and were plotted as the total infused FIX (x-axis) vs measured plasma FIX (y-axis). The empirically determined FIX concentrations were determined by ELISA (Methods).The plotted line is the fit of the experimental data (FIXt-C), where C is the calculated complex. The error bars represent the standard deviation of the mean for 8 or more mice for each level of infused FIX. The 2 light red points in the figure represent data collected in our laboratory about 5 years ago, and is only included as in internal check. The table above the figure represents the statistics of the quadratic fit.
Saphenous vein bleeding assay
The saphenous vein bleeding assay (SVBA) was used to assess hemostasis as previously described.23 FIX was infused into the left saphenous vein of hemophilia B mice and, after 7 days (in most cases), the right saphenous vein was used for the assay. In the SVBA, after initial vein injury, each time a clot forms it is mechanically disrupted, and the number of clots that occur over the course of 30 minutes recorded. An increased number of clots equates with better hemostasis.
Times to first clot
The right saphenous vein of CRM− mice was injured as described for the SVBA, and 15 minutes was allowed for any baseline clotting to occur. In CRM− mice, clot formation ceases by 8 minutes postinjury; any clots formed during the first 8 minutes were physically removed. Fifteen minutes after the initial injury, the different FIX agents were infused into the left saphenous vein and the time until the first clot formed at the right saphenous vein injury site, determined during a 20-minute window. There is insufficient blood loss during this time to affect clotting.
Activation of FIX molecules
FIX was activated with FXIa. To remove the FXIa activator, the generated FIXas were isolated by binding to a HiTrap Q column, washing with 250 mM salt, and eluting with 10 mM CaCl2 in 250 mM salt (method courtesy of Dougald Monroe, University of North Carolina).
Statistics
The Mann-Whitney test was used to compare SVBA results between groups. The statistics for samples comparing CRM+ and CRM− mice used a median test with P value derived by a Bootstrap method24 that adjusts for the different backgrounds. Except for the Bootstrap method, all statistical calculations were done with Mathematica, version 11.3.
Results
Estimation of vascular Col4 available for FIX binding
Previous work demonstrated that the rapid initial disappearance of FIXWT from plasma after infusion into a CRM− hemophilia B mouse6 is predominantly a result of its binding to extravascular Col4.10,11 There is a significant amount of Col4 in the body, but the amount capable of binding to FIX is unknown. We therefore estimated the amount of Col4 readily available for binding plasma FIX by infusing increasing doses of FIX (8-20 mice per dose) and taking plasma samples after 10 minutes for ELISA analysis. The following assumptions were made: a fraction of total body Col4 is readily accessible to the plasma;5,6 the binding of rFIX to Col4 has 1:1 stoichiometry, is reversible, and reaches near equilibrium by 10 minutes; the plasma volume in the mouse is 40 mL/kg21 ; and clearance of FIX from the plasma through other mechanisms other than by binding Col4 is negligible within the first 10 minutes after infusion.
Mathematica (version 11.3) NonLinearModelFit was used to fit the data to the quadratic equation for chemical equilibrium,22,25 and the total Col4 (Col4t) available for binding FIX and the in vivo value for Kd were estimated. The best least squares fits for Col4t and Kd were predicted to be 405 and 40 nM, respectively, with 95% confidence intervals of 342 to 467 and −7.1 to 87.0 nM (Figure 1). If, after simultaneous fitting of both parameters to the data, the result of Col4t is input into the quadratic equation and again fit to the data, the same parameters are achieved. However, P values and confidence intervals are much improved. For example, if 404 nM is input as Col4t, the predicted confidence interval for Kd is 16.5 to 63.5
Hemostatic efficacy of FIXAlb in CRM− mice
Recombinant FIX products with extended plasma half-lives are marketed to simplify prophylactic therapy in patients with hemophilia B. These molecules include FIXFC (Alprolix, a fusion of FIX and the Fc domain of immunoglobulin G)26 and FIXAlb (Idelvion, a fusion of FIX and albumin).27 We previously demonstrated that the hemostatic effects of FIXWT and FIXFC are equivalent in CRM− hemophilia B mice.11,12 Here, similar studies were performed using FIXAlb to test whether other longer circulating FIX’s behaved similarly. Similar to FIXFC, FIXAlb is not statistically superior to FIXWT in CRM− mice at any of the doses tested (Figure 2).
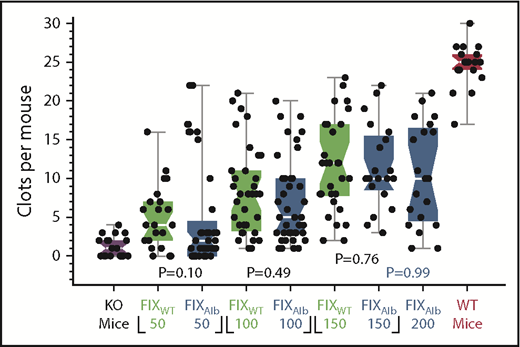
Comparison of prophylactic FIXWT and FIXAlb treatment in CRM− mice. SVBA 7 days after infusion of FIXWT (green) or FIXAlb (blue) into CRM− mice at 50, 100, 150,and 200 IU/kg. Results for untreated CRM− (purple) and WT (red) mice are also shown. There is no significant difference between the median SVBA values for the FIX agents. Box plot rendition of the results showing the minimum, first quartile, median (waist), third quartile, and maximum values. KO, knockout.
Comparison of prophylactic FIXWT and FIXAlb treatment in CRM− mice. SVBA 7 days after infusion of FIXWT (green) or FIXAlb (blue) into CRM− mice at 50, 100, 150,and 200 IU/kg. Results for untreated CRM− (purple) and WT (red) mice are also shown. There is no significant difference between the median SVBA values for the FIX agents. Box plot rendition of the results showing the minimum, first quartile, median (waist), third quartile, and maximum values. KO, knockout.
Effect of endogenous, dysfunctional (CRM+) FIX on hemostasis after FIXWT infusion in mice
The majority of patients with hemophilia B are CRM+; that is, they express a dysfunctional FIX protein. For example, 69 of the individuals with hemophilia B in the FIX variant database15 have the missense mutation R333Q. These patients generally have functional and antigenic levels of FIX in plasma of less than 3% and 100%, respectively, a moderate to severe bleeding diathesis, and a form of dysfunctional FIX that is expected to interact with Col4 normally. We hypothesized that although a dysfunctional FIX that binds Col4 would increase the plasma recovery of infused FIX, it would decrease the amount of infused FIX bound to Col4, and thereby potentially reduce hemostatic effectiveness.13 To test this hypothesis, hemostasis was compared 7 days after the infusion of FIXWT in CRM− and hFIX(R333Q) knock-in CRM+ hemophilia B mice. These CRM+ mice are reported to have a circulating hFIX(R333Q) plasma level of 1.2 µg/mL (21.4 nM), and similar to their human counterpart, a severe hemophilia phenotype.6 As predicted, the results (Figure 3) confirm a deleterious effect of the endogenous, dysfunctional FIX on hemostasis in CRM+ mice when compared with the same dose of FIXWT in CRM− mice (median SVBA values of 4 and 12, respectively; P < .001).
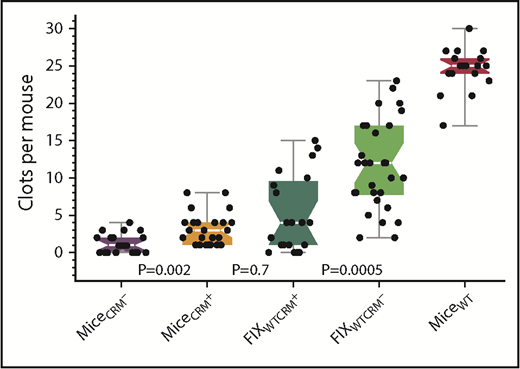
Reduced efficacy of prophylactic FIXWT treatment in CRM+ compared with CRM− mice. SVBA evaluation of hemostasis 7 days after the infusion of 150 IU/kg of FIXWT in CRM− and CRM+ mice. Baseline SVBA in untreated CRM− (black) and CRM+ (orange) mice, CRM+ mice infused with FIXWT (green), CRM− mice infused with FIXWT (light green), and wild-type mice (red). Box plot rendition of the results showing the minimum, first quartile, median (waist), third quartile, and maximum values.
Reduced efficacy of prophylactic FIXWT treatment in CRM+ compared with CRM− mice. SVBA evaluation of hemostasis 7 days after the infusion of 150 IU/kg of FIXWT in CRM− and CRM+ mice. Baseline SVBA in untreated CRM− (black) and CRM+ (orange) mice, CRM+ mice infused with FIXWT (green), CRM− mice infused with FIXWT (light green), and wild-type mice (red). Box plot rendition of the results showing the minimum, first quartile, median (waist), third quartile, and maximum values.
Comparison of FIXWT, FIXFC, and FIXAlb in CRM+ mice
Figure 4A shows that 7 days after infusion of 150 IU/kg (340 nM) into the CRM+ mice, FIXWT, FIXFC, FIXAlb did not produce SVBA results significantly different from those of untreated CRM+ mice. In addition, FIXFC and FIXAlb were not statistically different from FIXWT, nor were they different from each other. The observation that 6 of the 20 CRM+ mice infused with FIXFC had SVBA results within the range for WT mice is notable, suggesting that FIXFC might be superior to the other FIX molecules in CRM+ mice. This led us to investigate whether the potential benefits of FIXFC or FIXAlb, compared with FIXWT, would become apparent at higher doses in CRM+ mice. Figure 4B shows that hemostasis in CRM+ mice 7 days after infusion of very high doses of FIXFC (500 IU/kg) was significantly better than that in untreated CRM+ mice (median SVBA, 10.5 and 3.0, respectively; P = .014). Infusions of FIXWT (800 IU/kg) or FIXAlb (500 IU/kg), however, did not produce results significantly better than the CRM+ mouse background; moreover, the results with FIXWT and FIXAlb were not statistically different from one another (P = .38). Thus, of the 2 longer circulating FIXs tested, only FIXFC appears to be marginally superior to FIXWT in CRM+ mice.
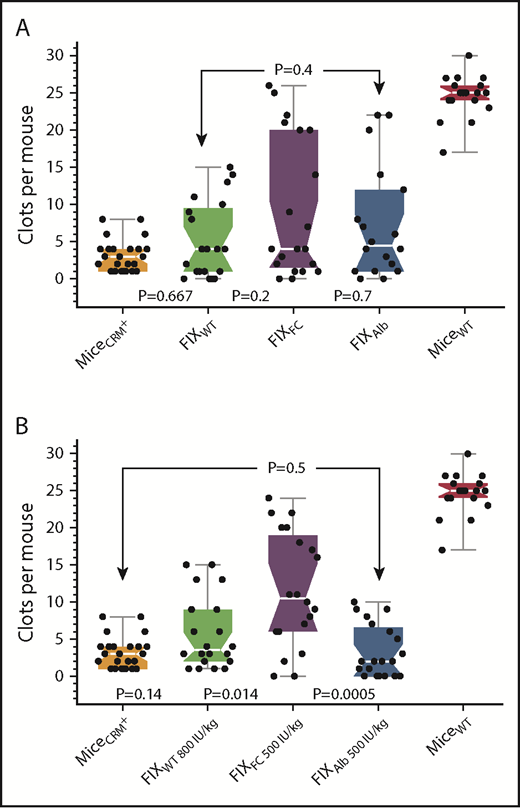
Comparison of prophylactic treatment with the different FIX agents in CRM+ mice. (A) SVBA 7 days after the infusion of 150 IU/kg FIXWT, FIXFC, or FIXAlb in CRM+ mice. Results for untreated CRM+ and wild-type mice are also shown. (B) SVBA 7 days after the infusion of extremely high doses of FIXWT, FIXFC, and FIXAlb in CRM+ mice. Untreated CRM+ mice (orange), FIXWT (green), FIXFC (purple), FIXAlb (blue), WT mice (red). Box plot rendition of the results showing the minimum, first quartile, median (waist), third quartile, and maximum values. Note no difference between agents at the 150 IU/kg dose, but significantly increased efficacy with FIXFC at very high doses.
Comparison of prophylactic treatment with the different FIX agents in CRM+ mice. (A) SVBA 7 days after the infusion of 150 IU/kg FIXWT, FIXFC, or FIXAlb in CRM+ mice. Results for untreated CRM+ and wild-type mice are also shown. (B) SVBA 7 days after the infusion of extremely high doses of FIXWT, FIXFC, and FIXAlb in CRM+ mice. Untreated CRM+ mice (orange), FIXWT (green), FIXFC (purple), FIXAlb (blue), WT mice (red). Box plot rendition of the results showing the minimum, first quartile, median (waist), third quartile, and maximum values. Note no difference between agents at the 150 IU/kg dose, but significantly increased efficacy with FIXFC at very high doses.
Time course of the hemostatic effects of FIXWT and FIXFC in CRM− and CRM+ mice.
After the infusion of 150 IU/kg of FIXWT or FIXFC, the SVBA was used to assess hemostasis at 1 and 7 days in CRM− mice (Figure 5A), and at 1, 3, 5, and 7 days in CRM+ mice (Figure 5B). In Figure 5C, the median SVBA results in Figure 5A-B, after adjustment for the backgrounds in untreated mice, are shown.
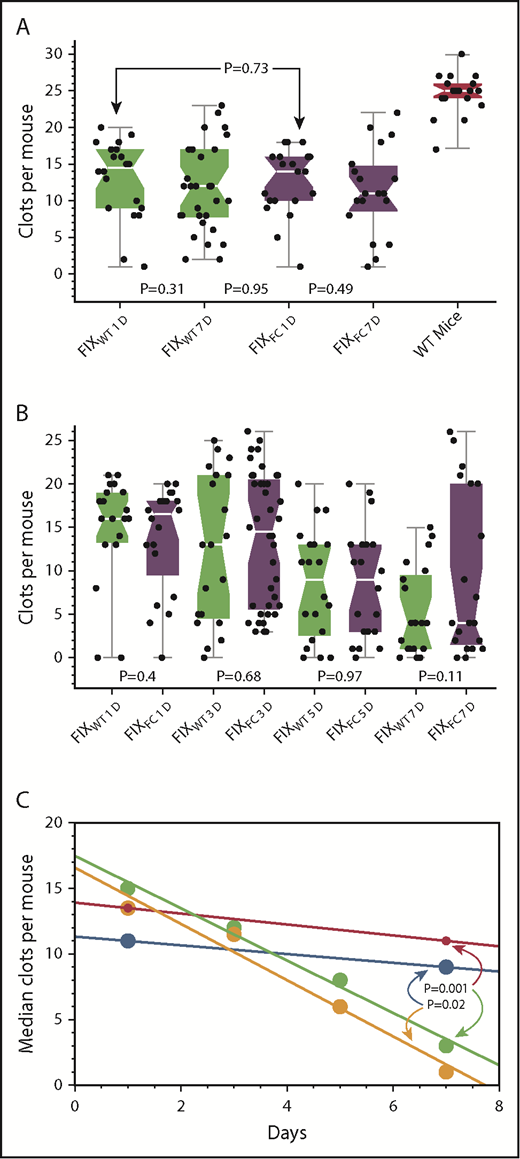
Time course of hemostasis after infusions of FIXWT and FIXFC in CRM− and CRM+ mice. (A) SVBA at 1 and 7 days after the infusion of 150 IU/kg FIXWT (green) or FIXFC (purple) in CRM− mice. (B) SVBA at 1, 3, 5, and 7 days after the infusion of 150 IU/kg FIXWT (green) or FIXFC (purple) in CRM+ mice. (C) Plot of median SVBA values for FIXWT (red) and FIXFC (blue) in CRM− mice and for FIXWT (green) and FIXFC (orange) in CRM+ mice from panels A and B after adjustment for the median SVBA baseline values of untreated CRM− (1) and CRM+ (3) mice. Note no significant difference in the hemostatic effects of the agents in CRM+ or CRM− mice at 24 hours, but the rate of decay in hemostatic effectiveness is much faster in CRM+ compared with CRM− mice. At 7 days, the differences between SVBA results in CRM− and CRM+ mice are statistically significant for both FIXWT (P=.001) and FIXFC (P = .02) treatments.
Time course of hemostasis after infusions of FIXWT and FIXFC in CRM− and CRM+ mice. (A) SVBA at 1 and 7 days after the infusion of 150 IU/kg FIXWT (green) or FIXFC (purple) in CRM− mice. (B) SVBA at 1, 3, 5, and 7 days after the infusion of 150 IU/kg FIXWT (green) or FIXFC (purple) in CRM+ mice. (C) Plot of median SVBA values for FIXWT (red) and FIXFC (blue) in CRM− mice and for FIXWT (green) and FIXFC (orange) in CRM+ mice from panels A and B after adjustment for the median SVBA baseline values of untreated CRM− (1) and CRM+ (3) mice. Note no significant difference in the hemostatic effects of the agents in CRM+ or CRM− mice at 24 hours, but the rate of decay in hemostatic effectiveness is much faster in CRM+ compared with CRM− mice. At 7 days, the differences between SVBA results in CRM− and CRM+ mice are statistically significant for both FIXWT (P=.001) and FIXFC (P = .02) treatments.
The hemostatic effects of FIXWT and FIXFC were not statistically different at any point in either the CRM− or CRM+ mice (Figure 5A and 5B). As assessed by the SVBA median results, however, the decline in the protective hemostatic effect of the infusions was much faster in CRM+ than in CRM− mice (Figure 5C). At 7 days after infusion, the difference between the results in CRM− and CRM+ mice was statistically significant for both FIXWT (P = .001) and FIXFC (P = .02) treated mice.
Effect of FIX reagents on an extant hemorrhage
The various FIXs were infused into CRM− mice 15 minutes after saphenous vein injury to determine the time necessary for the first clot to form in an ongoing hemorrhage (Figure 6). The mean times to first clot (TTFC) were 5.9, 7.3, and 11.8 minutes for FIXWT, FIXFC, and FIXAlb, respectively, with the TTFCs for both FIXFC and FIXAlb significantly prolonged compared with that for FIXWT (P = .007 and P < .001, respectively). FIXAlb displayed extreme variation in its TTFC; indeed, 3 of 15 mice infused with FIXAlb failed to clot during the 20-minute observation period.
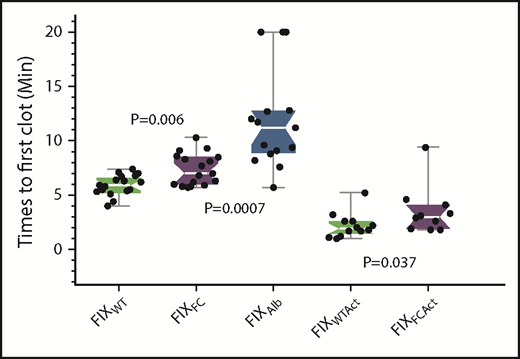
Times to first clot for the different FIX agents in CRM− mice. The saphenous vein of CRM− mice was injured and the mice were allowed to bleed for 15 minutes. Each FIX agent (500 IU/kg) was then infused and the TTFC (see Methods) determined. FIXWT (green), FIXFC (purple) and FIXAlb (blue). FIXWTAct and FIXFCAct are the activated forms of the respective zymogens. Box plot rendition of the results showing the minimum, first quartile, median (waist), third quartile, and maximum values.
Times to first clot for the different FIX agents in CRM− mice. The saphenous vein of CRM− mice was injured and the mice were allowed to bleed for 15 minutes. Each FIX agent (500 IU/kg) was then infused and the TTFC (see Methods) determined. FIXWT (green), FIXFC (purple) and FIXAlb (blue). FIXWTAct and FIXFCAct are the activated forms of the respective zymogens. Box plot rendition of the results showing the minimum, first quartile, median (waist), third quartile, and maximum values.
Part of the delay in TTFC may be explained by the time required to activate each zymogen molecule. Therefore, the TTFC assay was repeated with the activated proteins FIXWTAct and FIXFCAct. (The creation of FIXAlbAct is not possible because the C-terminal albumin is proteolytically removed during activation.) TTFCs were significantly faster for both activated FIXWT and FIXFC (1.9 and 2.9 minutes, respectively) than their zymogen forms, but the TTFC for activated FIXFC was still significantly slower than that for activated FIXWT (P = .037).
Discussion
Large amounts of Col4 are present in the body, but not all Col4 binds FIX. For example, despite its access to Col4 in the liver sinusoids, FIX is not bound.12 Presumably, lack of FIX binding to the Col4 in the liver sinusoids is the result of an unknown posttranslational modification of the Col4, rendering it unable to bind FIX. One example could be prolyl hydroxylation.28,29 Most of the subendothelium, however, contains Col4 that binds FIX avidly, thus producing a cache of FIX in basement membranes that is severalfold greater than the FIX circulating in plasma. That this FIX-Col4 complex plays an essential role in hemostasis is best illustrated by the bleeding diathesis evident in knock-in mice expressing FIXK5A, which has normal in vitro coagulant activity, but binds Col4 poorly.13,30
Our studies suggest the concentration of extravascular Col4 capable of binding plasma FIX in the mouse is 405 nM with a 95% confidence interval of 342 to 467 nM. This value is consistent with previously reported studies examining the competitive release of endogenous extravascular FIX in baboons.5 Binding to Col4 also appears to explain the 40% to 50% expected recovery of infused FIX observed in patients with hemophilia B.7 The majority of patients with hemophilia B are CRM+ and have variable circulating levels of a dysfunctional FIX. Depending on the level of the dysfunctional FIX and its ability to bind Col4, this CRM+ FIX could increase the plasma recovery of infused FIXWT, but reduce the quantity of FIXWT bound to vascular Col4 by competing for Col4 binding.
The Gla domain of FIX is responsible for its binding to Col4. For example, targeted mutation of 6 specific residues within the Gla domain of FVII is sufficient to produce a molecule with the same Kd for Col4 as FIXWT. Further, replacing the Gla domain of protein C with that of FIX produces molecules that binds Col4 with the same Kd as FIX.9,31 The majority of the described missense mutations in CRM+ patients with hemophilia B should not affect the sequence15 or function32 of the Gla domain and will, therefore, produce FIX proteins that presumably compete with infused FIX for Col4 binding. To examine coagulation in a situation mimicking prophylactic therapy, we tested hemophilia B mice with the SVBA assay 7 days after the infusion of the FIX. Hemostasis after the injection of FIXWT was substantially better in CRM− than in CRM+ (FIXR333Q) mice (Figure 3). Under the same experimental conditions, infusion of FIXAlb into either CRM− or CRM+ mice produced results indistinguishable from those after the infusion of FIXWT, including the reduction in hemostasis found in CRM+ compared with CRM− recipients (Figure 4A-B).
We had previously demonstrated that infusions of FIXWT and FIXFC also produced equivalent hemostatic results in CRM− mice, using this same model.12 At extremely high levels of FIX infusion (≥500 IU/kg), however, enhanced hemostasis is apparent when FIXFC is compared with FIXWT and FIXAlb in CRM+ mice (Figure 4B). The longer plasma half-life of FIXFC may contribute to its improved effect over FIXWT at these high levels of the infused FIX. The reason for the disparity in the results between FIXFC and FIXAlb, another long half-life agent, however, is not apparent (see the following paragraph).
The hemostatic effects measured with the SVBA are remarkably preserved 7 days after the infusion of FIXWT into CRM− mice, when plasma levels are not measurable11 (Figure 5A), consistent with a functional role for the FIX-Col4 vascular complex in hemostasis. In contrast, we observe a progressive decrease in hemostatic effectiveness during the same period in CRM+ mice (Figure 5B). The differences in hemostatic efficacy when the mouse has endogenous FIX may reflect the continuing expression of dysfunctional FIX, which competes for Col4 binding, in the CRM+ animals. Interestingly, we observe a linear kinetics loss of hemostatic protection over time (Figure 5C). This linear loss of efficacy contrasts with the well-documented exponential kinetics loss of infused FIX from the plasma of many animals.6,32,33 This suggests that hemostatic protection is attributable to something other than simply the plasma level of FIX and is consistent with our argument that extravascular FIX plays a functional role in hemostasis. Thus, in a CRM+ background because of a dysfunctional FIX that binds Col4, treatment with a FIX molecule with enhanced Col4 binding such as FIXK5R11 might be advantageous.
To evaluate further the different FIX molecules, their hemostatic effectiveness was tested in bleeding CRM− mice as the TTFC. Mouse blood completely circulates about 6 times per minute, and an inert molecule is mixed entirely by 25 seconds.34 Compared with FIXWT, the TTFC of both longer half-life FIX molecules is significantly delayed (Figure 6). The cause or causes of the apparent slower in vivo onset of coagulation of the zymogen FIXFC and FIXAlb forms in the mouse are likely multifactorial and could include differences in their rates of activation, the activities of their activated FIXa forms once generated, and perhaps their binding to Col4. In the latter regard, it is of interest that dosing recommendations in the package inserts of BeneFix (FIXWT), Alprolix (FIXFC), and Idelvion (FIXAlb) suggest plasma recoveries of the infused products to be 38%, 50%, and 65%, respectively. Given the dominant effect of vascular Col4 binding on the plasma recovery of the infused FIX, the dosing recommendations suggest that the relative degree of Col4 binding of the agents is FIXWT > FIXFC > FIXAlb. Of the longer half-life molecules, the efficacy of FIXFC appeared to be superior to that of FIXAlb in our mouse hemophilia B models.
On the basis of plasma FIX levels, the longer half-life FIX products are administered at a reduced frequency (1/wk) for prophylactic treatment and were rapidly accepted by the hemophilia B community. Studies supporting the efficacy of the less frequent prophylactic doses of these products, however, do not consider the potential procoagulant effect of the extravascular FIX bound to Col4. FIXWT, FIXFC, and FIXAlb all produce equivalent results in our mouse model of once-per-week prophylactic therapy, but clinical studies directly comparing the various FIX agents have not been performed. Of note in this regard, once-per-week dosing of 100 IU/kg BeneFix (FIXWT) in patients with hemophilia B appears to produce outcomes similar to those reported for Alprolix (FIXFC).35
We believe the deleterious hemostatic effect of the dysfunctional FIX tested in our mouse studies (FIXR333Q) is primarily a result of its competition with infused FIX for Col4 binding. FIX bound to Col4 in the extracellular matrix of the subendothelium is in an ideal location to contribute to the hemostatic response to vascular injury. Tissue factor, which is responsible for the initiation of coagulation, is expressed by pericytes, which share the extracellular matrix of microvascular endothelial cells.36 Plasma von Willebrand factor carries FVIII and also binds Col4 specifically.36,37 Further, platelets bind collagens of various types, including Col4, so the possibility that all these procoagulant elements combine locally on a subendothelial scaffold to augment clotting is appealing.
It must be pointed out, however, that the endogenous expression of a dysfunctional FIX could affect hemostasis through other mechanisms as well, including competition with FIX for activation by FVIIa/tissue factor or FXIa and competition with FIXa for FVIIIa binding. The latter mechanism is unlikely in our tested CRM+ mouse, as the hemostatic defect with FIXR333Q is a result of its much-reduced affinity for FVIIIa.38
The presence of CRM+ material may explain some of the variability in disease severity in patients with hemophilia B with the same level of plasma FIX functional activity. For example, a patient whose FIX is 100% active with a plasma antigen level of only 2% may have very different bleeding expectations than a CRM+ patient expressing a dysfunctional FIX with 2% activity and an antigen level of 100%.
In sum, these studies in murine hemophilia B models suggest a potential, unappreciated role of the endogenous, dysfunctional FIX in CRM+ individuals in attenuating their hemostatic response to prophylactic therapy. The extent of the deleterious effect would depend on the level of expression of the dysfunctional FIX and its specific molecular abnormality.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Katherine Stafford and Sheue-Mei Wu for editing and discussions.
This work was supported by a trust fund of D.W.S.
Authorship
Contribution: D.W.S. originated the work and wrote the first draft; D.W.S. and D.M.M. planned most of the experiments; B.C. performed the experiments and helped plan the time to first clot experiments; G.J.B. analyzed the data and helped write (and rewrite) the manuscript; F.-C.L. evaluated most of the data and performed some of the statistical calculations; L.G.P. was also involved in planning and especially spent hours understanding and helping with the mathematical evaluations and also spent considerable time with revisions; and all authors carefully read the final version and made helpful comments.
Conflict-of-interest disclosure: D.W.S. has a patent on the VKOR enzyme and, in Europe, on FIXK5R/R338L (the latter is under consideration in the United States). The remaining authors declare no competing financial interests.
Correspondence: Darrel W. Stafford, Department of Biology and Pathology, University of North Carolina at Chapel Hill, 442 Wilson Hall, South Road, Chapel Hill, NC; e-mail: dws@e-mail.unc.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal