

Abstract
RNA-binding proteins (RBPs) regulate fundamental processes, such as differentiation and self-renewal, by enabling the dynamic control of protein abundance or isoforms or through the regulation of noncoding RNA. RBPs are increasingly appreciated as being essential for normal hematopoiesis, and they are understood to play fundamental roles in hematological malignancies by acting as oncogenes or tumor suppressors. Alternative splicing has been shown to play roles in the development of specific hematopoietic lineages, and sequence-specific mutations in RBPs lead to dysregulated splicing in myeloid and lymphoid leukemias. RBPs that regulate translation contribute to the development and function of hematological lineages, act as nodes for the action of multiple signaling pathways, and contribute to hematological malignancies. These insights broaden our mechanistic understanding of the molecular regulation of hematopoiesis and offer opportunities to develop disease biomarkers and new therapeutic modalities.
Introduction
RNA may code for protein (messenger RNA [mRNA]), but it also performs functions exemplified by ribosomal RNA, microRNA, transfer RNA, and long noncoding RNAs. The biogenesis, fate, and function of these molecules are directed by dynamic interactions with RNA-binding proteins (RBPs). More than 1500 proteins have been annotated as RBPs, based upon the presence of characteristic RNA binding domains or their residence within established ribonuclear protein complexes.1 Experimental approaches using UV cross-linking of RNA to protein, followed by selection of polyadenylated RNAs and analysis of bound proteins by mass spectroscopy, revealed that many RBPs lacked any conventional RNA binding domain and had no previous connection to RNA biology.2-4 Among these unexpected RBPs was an enrichment for metabolic enzymes. The exact nature and functional consequences of many of these interactions remain unclear. A general assumption is that RBPs regulate the fate of the bound mRNA; however, in some instances, the mRNA may regulate the protein.
More than 40 RNA binding domains have been described. Although each recognizes relatively common minimal elements, specificity is enhanced by the combinatorial use of multiple domains. Regulatory cis-elements, bound by RBPs, are typically located within 5′ or 3′ untranslated regions (UTRs). These elements can be extremely diverse and may include short sequence motifs, simple hairpin-like structural elements, or highly complex folded structures. RBPs regulate mRNA 5′ capping, splicing, polyadenylation, nuclear export, localization, translation, silencing, and decay, thereby generating diversity in the expressed transcriptome and proteome. The ability of RBPs to be controlled by posttranslational modifications affords a mechanism whereby the cellular transcriptome and proteome can be rapidly remodeled. A single RBP may regulate a cohort of transcripts that affect a common process, a so-called “RNA regulon.”5,6 The use of next-generation sequencing technologies has increasingly highlighted the importance of RBPs, as well as their roles in splicing and translation, in the pathogenesis of specific hematological malignancies. In this review, we focus on how RBPs regulate mRNA, emphasizing their role in hematopoiesis and hematological malignancy, and the implications for potential therapy.
Splicing
Almost all mRNAs are transcribed as a pre-mRNA containing introns that are excised by a multiprotein complex known as the spliceosome (Figure 1A). Most human genes give rise to alternatively spliced transcripts,7,8 and these isoforms may show cell-type specificity. Alternative splicing may affect the proteome qualitatively by generating variant protein isoforms from the same gene. It may also regulate the quantity and timing of protein expression, through intron retention, which has emerged from studies of granulopoiesis and erythropoiesis as a common and developmentally regulated mechanism of gene expression.9,10 Although some retained introns may promote nuclear transcript decay or non-sense–mediated decay (NMD), others result in nuclear retention and resistance to NMD, with subsequent splicing and protein expression delayed until a later developmental stage.11,12
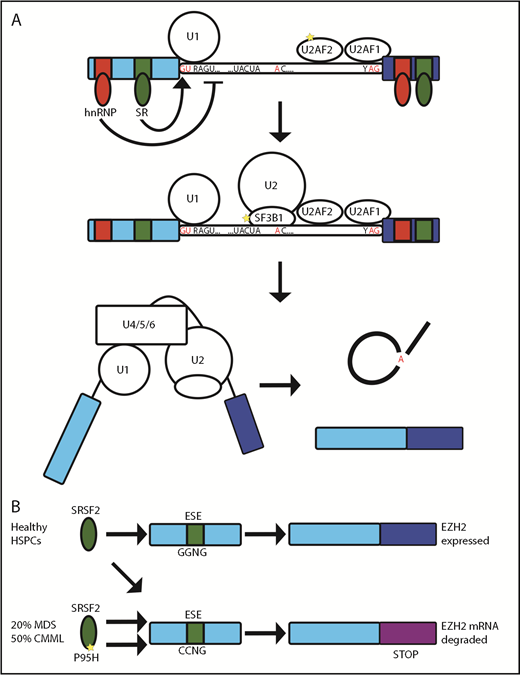
SF3B1, SRSF2, and U2AF function in splicing. (A) The U1 small nuclear ribonucleoprotein (snRNP) and U2AF initially bind to the 5′′ and 3′′ splice sites, respectively. This is followed by binding of the SF3B-containing U2 snRNP and, subsequently, assembly of a multiprotein complex (including U4, U5, and U6 snRNPs) known as the “spliceosome,” which then leads to excision of the intervening intron. Further sequences within exons and introns act as splicing enhancer or silencer elements and are bound by proteins, such as hnRNPs and SR proteins (eg, SRSF2). These RBPs allow splicing to be controlled in a tissue-developmental stage– and stimulus-specific manner. (B) SRSF2 binds equally to GGNG and CCNG exonic splicing enhancers (ESE) to allow expression of EZH2 in healthy HSPCs. In MDS/chronic myelomonocytic leukemia, the P95H mutation of SRSF2 has preferential binding to the CCNG ESE, giving rise to a splice variant of EZH2 including an exon with a premature stop codon that is degraded by NMD.
SF3B1, SRSF2, and U2AF function in splicing. (A) The U1 small nuclear ribonucleoprotein (snRNP) and U2AF initially bind to the 5′′ and 3′′ splice sites, respectively. This is followed by binding of the SF3B-containing U2 snRNP and, subsequently, assembly of a multiprotein complex (including U4, U5, and U6 snRNPs) known as the “spliceosome,” which then leads to excision of the intervening intron. Further sequences within exons and introns act as splicing enhancer or silencer elements and are bound by proteins, such as hnRNPs and SR proteins (eg, SRSF2). These RBPs allow splicing to be controlled in a tissue-developmental stage– and stimulus-specific manner. (B) SRSF2 binds equally to GGNG and CCNG exonic splicing enhancers (ESE) to allow expression of EZH2 in healthy HSPCs. In MDS/chronic myelomonocytic leukemia, the P95H mutation of SRSF2 has preferential binding to the CCNG ESE, giving rise to a splice variant of EZH2 including an exon with a premature stop codon that is degraded by NMD.
Splicing isoform usage is regulated in a tissue-, developmental stage–, and stimulus-specific manner, often by posttranslational modification of RBPs. An example is the RBP heterogeneous nuclear ribonucleoprotein A1 (hnRNPA1); among its many contributions to RNA metabolism,13 hnRNPA1 controls splicing of transcripts required for hematopoiesis. In turn, hnRNPA1 is regulated by the ubiquitin ligase TRAF6 downstream of Toll-like receptor signaling.14 Enforced TRAF6 ubiquitination of hnRNPA1 in mouse hematopoietic stem/progenitor cells (HSPCs) led to aberrant splicing and bone marrow failure. This link among Toll-like receptor signaling, splicing, and bone marrow failure is of particular interest because of the relationship between chronic inflammation and disorders such as myelodysplasia.15 There is considerable evidence that alternative splicing plays an important role in the pathogenesis of myelodysplasia and other hematological malignancies. Missense mutations in splicing factors are found in >50% of cases of myelodysplastic syndrome (MDS).16-18 They are also commonly identified in clonal hematopoiesis19,20 and CLL.21,22 Although the spliceosome includes >200 individual proteins, recurrent mutations are restricted to SRSF2, SF3B1, U2AF1, and ZRSR2. Interestingly, these mutations are always hemizygous missense mutations and are mutually exclusive. The apparent requirement to retain 1 wild-type allele might be therapeutically exploitable, and mouse models of splice factor mutation leukemia suggest an increased sensitivity to inhibitors of the spliceosome.23
SRSF2 is a splicing factor that promotes exon recognition by the binding of its RNA recognition motif to exonic splicing enhancer sequences in pre-mRNA to recruit further components of the spliceosome. SRSF2 is mutated in >20% MDS and 50% chronic myelomonocytic leukemia, with P95H being the most common mutation. A heterozygous mouse model of the Srsf2 P95H mutation developed an expanded hematopoietic progenitor compartment with increased proliferation, apoptosis, and peripheral blood cytopenias reminiscent of human MDS.24 In contrast, hematopoietic failure was seen after either homozygous deletion or monoallelic expression of mutant Srsf2 confirming the requirement to retain 1 wild-type allele.23,24 Mutation of P95 changed the RNA binding preference of SRSF2 in mouse and human cells resulting in an altered pattern of splicing that partially overlapped between studies and species.24-29 Ezh2 was identified as an aberrantly spliced transcript that was degraded by NMD, thereby reducing protein expression24 (Figure 1B). Furthermore, the hematopoietic phenotype was partially reversed by forced expression of Ezh2.24 However, aberrant splicing of Ezh2 could not be detected in 2 subsequent Srsf2 P95 mutant mouse models.27,29,30 Attempts to characterize the alternatively spliced transcriptome in human patients have included a comprehensive analysis of purified CD34+ HSPCs from patients with splicing factor mutant myelodysplasia. This identified many aberrant splicing events, with different mechanisms of altered splicing seen with each mutant splice factor. Although little overlap was observed at the individual gene level there was convergence onto common pathways.26
The splicing factor SF3B1 is part of the U2 small nuclear ribonucleoprotein that binds to the branchpoint sequence. The K700E mutation is common in MDS and chronic lymphocytic leukemia (CLL). Transcriptomic analysis of CLL patients has identified multiple programs dysregulated in the presence of the mutation,31 but whether this is the mechanism by which mutant SF3B1 contributes to CLL pathogenesis remains uncertain. Conditional knock-in of this mutation in mouse hematopoietic stem cells resulted in anemia and reproduces the broad picture of splicing alteration seen in human mutant myelodysplasia.32 However, the abnormally spliced transcripts showed almost no overlap between human and mouse, presumably as a result of the limited interspecies conservation of intronic sequences. The fact that phenotypes of abnormal hematopoiesis can be reproduced across mouse models of different mutant splicing factors, despite the limited overlap in the transcripts altered between human and mouse, suggests that it may be the global, rather than gene-specific, alteration in splicing that contributes to pathogenesis. Recently, more general effects of aberrant splicing, including R-loop formation and induction of the DNA damage response, have been suggested as contributory mechanisms.33
Spliceosome function may be altered, even in the absence of mutations in its protein constituents. Proteomic analysis showed increased expression of spliceosome components in CLL compared with normal B cells, even in the absence of splicing factor mutation, suggesting the fundamental importance of splicing in CLL.34 Consistent with the possibility of a generalized splicing defect, exposure to the SF3B1 inhibitor spliceostatin A induced apoptosis of CLL, but not normal B cells, independent of SF3B1 mutation status.35 SF3B1 inhibition with the drug E7107 synergized with, and was able to overcome resistance to, the BCL2 inhibitor venetoclax in the TCL1 mouse model of CLL and in human CLL cells.36 This effect was potentially due to splicing changes in BCL2 family genes, in particular MCL1. Importantly, efficacy did not require the presence of splice factor mutations, consistent with aberrant splicing as a generalized feature of CLL. These results are especially exciting, because E7107 has already entered clinical trials for patients with solid organ malignancies. However, although effects on splicing of predicted target genes were confirmed at the administered doses, the development of unexpected optic neuritis and visual loss led to study discontinuation on safety grounds.37,38 This toxicity, which had not been predicted from animal studies, emphasizes the need for a deeper understanding of the mechanism of action of these drugs and their target RBPs before these agents can be optimally deployed in humans.
Mutations may also be found in cis-regulatory elements that recruit the RBPs that regulate splicing. An example in CLL is NOTCH1, where mutation of a cryptic splice site generates a hyperstable form of NOTCH1.39 Similar mutations may have been dismissed as silent mutations or may be located in noncoding regions; as such, their significance may be underappreciated. Altered splicing may contribute to resistance to therapies. For example, loss of CD19 expression during chimeric antigen receptor T-cell therapy in acute lymphocytic leukemia is caused by altered expression of the RBP SRSF3 and consequent altered splicing of CD19 mRNA.40 New technologies for the qualitative analysis of RNA, including novel isoform identification, will facilitate a greater understanding of mRNA splicing control in normal and malignant hematopoiesis.
Polyadenylation
Qualitative changes in the transcriptome are also generated by alternative polyadenylation (APA). Most eukaryotic genes contain >1 polyadenylation signal sequence and, therefore, have the potential to express alternative 3′UTRs.41 Early observations described how cancer was broadly associated with APA and a global shortening of 3′UTRs, consistent with a generalized escape from posttranscriptional regulation.42 The selection of polyadenylation sites is influenced in a dynamic fashion by RBPs with ∼20 core proteins acting in complexes, such as the cleavage and polyadenylation specific factor, cleavage factor-1 and -2, poly(A) polymerase, and poly(A) binding proteins.43
APA may also occur at polyadenylation signals within intronic regions of the transcript. This can lead to exclusion of coding regions and the generation of truncated proteins with lost or altered function. Recent studies show that intronic polyadenylation is commonplace in normal and malignant cells, with differential intronic polyadenylation associated with progression of B-cell development.44 Furthermore, analysis of CLL identified widespread intronic polyadenylation, resulting in truncation of transcripts encoding potential tumor suppressors.45 Indeed, inactivation of potential tumor suppressors by intronic polyadenylation was more common than inactivation by DNA mutation. Altered transcripts included genes annotated as tumor suppressors in other malignancies but not previously recognized as being altered at the DNA level in CLL. Because APA will not be detected by genomic DNA sequencing, this suggests the existence of a dominant mechanism of tumor suppression that has yet to be fully explored. How the expression, mutation, and modification of RBPs contribute to the selection of APA sites during hematopoiesis and leukemogenesis is an exciting area of ongoing research.
APA may affect gene expression by several mechanisms. The simplest explanation is that shortening of the 3′UTR leads to the loss of cis-regulatory elements that determine binding of RBPs and microRNAs, in turn leads to altered expression of that transcript. An intriguing additional possibility arises from the suggestion that some transcripts act as sponges to sequester RBPs or microRNAs that would otherwise regulate the expression of additional transcripts, thereby acting as “competing endogenous RNAs.” A recent model-based analysis suggests that the regulation of many tumor suppressor transcripts is influenced by the APA and loss of regulatory elements in other competing endogenous RNA transcripts.46 This emphasizes the complexity of the networks of posttranscriptional regulation that are coordinated by RBPs.
Translation
The rate of translation is tightly controlled in normal hematopoiesis47 and commonly dysregulated in cancers, including hematological malignancies. Regulation of translation can occur during the initiation, elongation, and termination phases; however, initiation is often the rate-limiting step. Binding of the mRNA 5′cap by EIF4E is required for translation of most mRNAs. EIF4E forms part of the EIF4F complex with EIF4G and EIF4A. EIF4G acts as scaffold by binding to the 40S ribosome–containing preinitiation complex. EIF4A is a DEAD box containing helicase that unwinds structural elements in the 5′UTR as the preinitiation complex scans along 5′UTR until the 60S ribosome is recruited at a suitable start codon, and protein synthesis is initiated.
EIF4E overexpression is commonly seen in human cancer, and its forced expression can be transforming in vitro.48,49 In vivo experiments revealed that overexpression of EIF4E in transgenic mice led to the development of multiple cancer types, including B-cell lymphomas,50 and accelerated lymphomagenesis in a c-myc mouse model.51 The transforming effect of EIF4E was not due to the global increase in protein synthesis but rather to a previously recognized selectivity for transcripts related to proliferation, survival, and metabolism.49 Further evidence of this oncogenic specificity came from Eif4e+/− mice, in which 50% expression of EIF4E was sufficient for normal levels of global translation, as well as normal development, but insufficient to permit HRAS-induced cellular transformation.52 This was due to reduced translation of a “regulon” of transcripts required for cellular transformation. Enhanced EIF4E binding to these transcripts was mediated by a C-rich motif in the 5′UTR. The existence of this differential requirement for EIF4E expression suggests the existence of a therapeutic window that might be exploited to suppress tumor growth. In addition to its role in translation initiation, EIF4E may play a separate role in the selective export of oncogenic mRNAs from the nucleus. Indeed, this activity has been successfully targeted in clinical trials of acute myeloid leukemia (AML) using the m7G-cap analog ribavirin.53 The selectivity of EIF4E in promoting the nuclear export and translation of MYC, BCL2, and BCL6 has been proposed as a therapeutic strategy to target double- and triple-hit diffuse large B-cell lymphoma.54
Another potential therapeutic target is the RNA helicase EIF4A. Exposure of leukemic cells to the EIF4A inhibitor silvestrol leads to translational downregulation of a program of G-quadruplex–containing transcripts enriched for oncogenes and showed significant activity against leukemic cell lines.55 Activity of EIF4A is positively regulated by EIF4B, a protein that is overexpressed in diffuse large B cell lymphoma.56 Increased EIF4B promotes translation of an oncogenic regulon that includes antiapoptotic and DNA repair proteins. RNA helicases represent a promising target for anticancer drug development, especially because many such inhibitors are already in development as antiviral agents.
In addition to canonical cap-dependent translation facilitated by EIF4E, it is clear that other proteins may act as cap binders. The multisubunit eIF3 complex acts as a bridge between the 40S ribosome and EIF4G. However, subunit EIF3d is responsible for m7G-cap recruitment in many mRNAs and may allow ongoing translation in the absence of EIF4E or during mTOR inhibition. Indeed, EIF3 appears to regulate the translation of specialized mRNA regulons involved in proliferation and apoptosis.57 This specificity relates to structural elements within the 5′UTR of specific mRNAs that permit EIF3d m7G-cap binding.58 Knockdown of EIF3D inhibits cell proliferation in a number of cancer cell lines, including AML.59
The activity of these protein complexes and, thus, initiation are regulated by signaling pathways (Figure 2). The best characterized of these is mTORC1, which phosphorylates and inactivates eIF4E-BP to release the cap-binding activity of EIF4E. mTORC1 can also induce the degradation of PDCD4, a negative regulator of EIF4A, through phosphorylation via S6 kinase. In CD4 T cells, engagement of the T-cell receptor triggers rapid mTOR activation and alters metabolism through the translation of a program of preexisting or “poised” mRNAs.60 Translation is also regulated by the B-cell receptor (BCR). In CLL, engagement of the BCR is shown to enhance global translation and the specific translational upregulation of MYC.61 Increased MYC promotes expression of the translational machinery, increasing ribosome production and activity. These changes appeared to depend upon enhanced expression of EIF4A and EIF4G and a reduction in PDCD4. The role of BCR signaling as a tractable therapeutic target to influence downstream RBP activity was supported by the ability of inhibitors of BCR kinases SYK and BTK to fully or partially reverse the effects on translation.
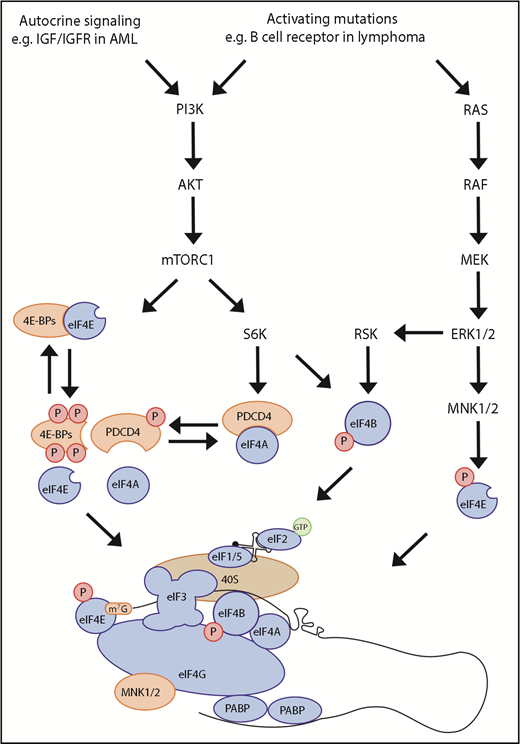
Signaling to cap-dependent translation initiation. Cap-dependent translation can be controlled through activation of the PI3K-mTOR and MAPK pathways. Binding of eIF4E and eIF4G is required for eIF4F function and translation of many mRNAs; however, this can be inhibited by eIF4E-binding protein (4E-BP). mTORC1 controls the binding of 4E-BP to eIF4E through phosphorylation of 4E-BP. Furthermore, mTORC1 can control the availability of eIF4A through activation of S6K1/2, which phosphorylates PDCD4, releasing eIF4A. Mitogen-activated protein kinase-interacting kinase 1/2, which is bound by eIF4G, can also regulate translation by phosphorylating eIF4E. The PI3K-mTOR and MAPK pathways converge to phosphorylate eIF4B, a cofactor of eIF4A, leading to increased eIF4A activity.
Signaling to cap-dependent translation initiation. Cap-dependent translation can be controlled through activation of the PI3K-mTOR and MAPK pathways. Binding of eIF4E and eIF4G is required for eIF4F function and translation of many mRNAs; however, this can be inhibited by eIF4E-binding protein (4E-BP). mTORC1 controls the binding of 4E-BP to eIF4E through phosphorylation of 4E-BP. Furthermore, mTORC1 can control the availability of eIF4A through activation of S6K1/2, which phosphorylates PDCD4, releasing eIF4A. Mitogen-activated protein kinase-interacting kinase 1/2, which is bound by eIF4G, can also regulate translation by phosphorylating eIF4E. The PI3K-mTOR and MAPK pathways converge to phosphorylate eIF4B, a cofactor of eIF4A, leading to increased eIF4A activity.
Further specificity of the translation initiation step is added by a number of mechanisms. The 5′UTRs of mRNAs encoding oncogenes may differ in length, structure, or specific sequence motifs, rendering them more sensitive to changes in the abundance or activity of RBPs. The terminal oligo-pyrimidine (TOP) motif is a regulatory element that is found almost exclusively in the 5′UTR of mRNAs encoding ribosomal subunits and components of the translational machinery. TOP motifs are bound by the RBP LARP1, which acts to suppress their translation. LARP1 is phosphorylated by mTORC1, leading to its dissociation and enhanced translation of TOP-containing transcripts.62 This provides a mechanism for mTOR signaling to coordinate increased global translation with increased ribosome biogenesis. Conversely, it provides a therapeutic opportunity to target RBPs and, thereby, translational activity, by inhibition of upstream signaling pathways, such as mTOR.
Musashi2 (MSI2) is an RBP with a role in promoting normal hematopoiesis and malignant transformation. MSI2 is highly expressed in hematopoietic stem cells (HSCs), but its expression declines at subsequent stages of myeloid differentiation. Knockdown in mouse progenitors was associated with reduced HSC numbers. Conversely, forced expression of MSI2 was associated with increased proliferation of HSCs and impaired myeloid differentiation. Consistent with these effects, MSI2 appears to play a proleukemic role in myeloid malignancies. Expression of MSI2 is increased in high-risk MDS and AML, where its increased expression correlates with poor prognosis.63,64 It binds directly to mRNAs encoding key transcriptional regulators of myelopoiesis, Hoxa9, Myc, and Ikzf2, and promotes their translation in a mouse model of AML. The expression of MSI2 is also increased in human chronic myeloid leukemia (CML) blast crisis and is required for CML transformation.65 Experiments in human CML cell lines and primary CML cells show that MSI2 binds the transcript encoding branched chain amino acid transaminase 1 and promotes its translation. Branched chain amino acid transaminase 1, itself required for CML transformation, is an enzyme that catalyzes the production of valine, leucine, and isoleucine and, thus, acts to enhance mTOR signaling and promote global translation initiation. These experiments suggest a critical leukemia-promoting role for MSI2 and potential as a therapeutic target. This is supported by the description of a small molecule that inhibits MSI–RNA interaction, which induced dose-dependent toxicity to AML cells in vitro and in vivo.66
A screen for MSI2-interacting partners that contribute to leukemogenesis in mice identified a second RBP, Syncrip (hnRNPQ1).67 Knockdown of Syncrip in mouse models of AML led to myeloid differentiation and apoptosis, whereas overexpression in a mouse AML cell line increased colony formation, enhanced cell growth, and accelerated leukemogenesis in an in vivo model. Analysis of transcriptomes showed that Syncrip cooperated with MSI2 to enforce the hematopoietic stem cell and leukemia stem cell state. Although not directly interacting with MSI2, Syncrip binds overlapping mRNA targets of MSI2, including Myc, Hoxa9, and Ikzf2, and it affects regulation, predominantly at the level of translation. Although Syncrip is essential for maintenance of malignant tumor cells, it is not required for normal murine hematopoiesis. The apparent selective requirement for Syncrip in malignant hematopoiesis is intriguing and suggests a potential therapeutic opportunity.
Although mechanisms that promote increased translation are generally associated with cellular transformation, there are also examples whereby suppressed translation contributed to malignant disease. Translational profiling in CLL showed a translational downregulation of ribosome protein subunits and translational suppression of dyskeratin, a protein required for ribosomal RNA processing and, hence, ribosome biogenesis. Indeed, those patients with reduced levels of dyskeratin were associated with comparatively reduced overall survival after chemotherapy.68 These and other observations suggest that tumor cells have a “sweet spot” of optimal translation and that perturbations from this optimum might stress or kill tumor cells.
mRNA methylation
Numerous chemical modifications to RNA have been described; however, the most prevalent and well studied is N6-methyladenosine (m6A).69 RNA methylation influences RNA fate in many ways, including effects on splicing, nuclear export, translation, and decay. The m6A modification is mediated by 2 methyltransferases, METTL3 and METTL14, and their cofactors WTAP, KIAA1429, and ZFP217. The m6A modification can be removed by demethylases, such as FTO and ALKBH5 (Figure 3A). Another set of proteins bind m6A and regulate transcript stability and translation. METTL3 may promote translation through an interaction with EIF3H that promotes mRNA looping and ribosome recycling from the stop codon. Disruption of this interaction suppresses the effect of METTL3 on translation and abolishes its ability to drive oncogenic transformation.70
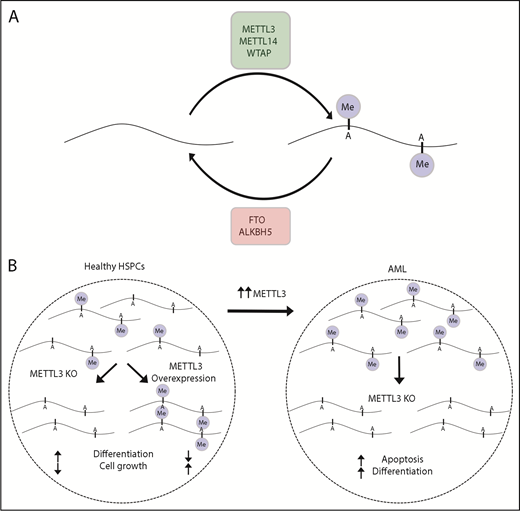
m6A mRNA methylation control and dysregulation in AML. (A) The amount of m6A mRNA methylation in a cell is determined by the activities of methyltransferases (METTL3 and METTL14) and demethylases (eg, FTO and ALKBH5). (B) In healthy HSPCs, knockout of METLL3 reduces methylation, increases differentiation, and reduces cell growth, whereas increased METTL3 has the opposite effect. AML cells frequently have increased METTL3 and increased methylation. In AML, knockout of METTL3 leads to increased differentiation and apoptosis.
m6A mRNA methylation control and dysregulation in AML. (A) The amount of m6A mRNA methylation in a cell is determined by the activities of methyltransferases (METTL3 and METTL14) and demethylases (eg, FTO and ALKBH5). (B) In healthy HSPCs, knockout of METLL3 reduces methylation, increases differentiation, and reduces cell growth, whereas increased METTL3 has the opposite effect. AML cells frequently have increased METTL3 and increased methylation. In AML, knockout of METTL3 leads to increased differentiation and apoptosis.
Regulation of m6A has recently emerged as a regulator of normal and malignant hematopoiesis.71,72 METTL3 and METTL14 are abundant in mouse and human hematopoietic stem cells and act to promote cell growth and repress differentiation. These proteins are also highly expressed in AML, and catalytically active METTL3 is required for maintenance of the malignant clone. Loss of METTL3 led to loss of m6A and diminished translation of components of oncogenic pathways, with an associated increased expression of genes involved in hematopoietic differentiation (Figure 3B). METTL3 also contributes to lymphoid homeostasis.73 When adoptively transferred to lymphopenic mice, Mettl3-deficient T cells were unable to expand or differentiate into effector cells, yet they persisted as naive T cells. This reflected a reduced ability of T cells to respond to interleukin-7 signaling as a result of increased expression of the METTL3 target transcripts SOCS1, SOCS3, and CISH. Although m6A is the best-studied RNA modification, it is clear that many other such modifications exist. How these modifications are imposed by protein effectors, how they influence RBP recruitment, and their involvement in hematopoiesis and hematological malignancy are likely to be unraveled in coming years.
mRNA decay
The cellular abundance of all RNA is determined by its rates of transcription and degradation. RNA decay is regulated by RBPs that recognize and bind to cis-regulatory RNA elements and recruit mediators of decapping, deadenylation, and exoribonucleolytic decay. A well-established element mediating decay is the AU-rich element. This is recognized by different RBPs, including the ZFP36 and ELAV families of RBPs. Mouse models provided the first evidence for the importance of these RBPs in the hematopoietic and immune systems. Zfp36l2-knockout mice showed depletion of hematopoietic stem cell and progenitor compartments and died within 2 weeks of birth from anemia and thrombocytopenia.74 The precise molecular mechanism was not established, but Zfp36l2 was proposed to balance HSC self-renewal with differentiation. Consistent with these findings, Zfp36l2 was shown to suppress a program of transcripts promoting erythroid differentiation.75 ZFP36L2 expression is decreased from the BFU-E stage onward, allowing expression of transcripts promoting terminal differentiation. Interestingly, despite sharing almost identical zinc finger RNA interaction domains, germline knockouts of the 2 other family members, Zfp36 and Zfp36l1, show very different phenotypes. A Zfp36 knockout displayed a proinflammatory phenotype due to increased stability and overexpression of cytokines, such as tumor necrosis factor, granulocyte-macrophage colony-stimulating factor, and interleukin-23, whereas the Zfp36l1 knockout had a lethal phenotype at embryonic day 9.5.76-78 These findings demonstrate an unexpected degree of specificity between seemingly similar RBPs that may relate to tissue-specific expression levels, tissue-specific posttranscriptional regulation, or the ability to recruit effector proteins.
More recently, conditional mutant mouse models have revealed roles for these proteins in hematopoiesis and the immune system. Simultaneous deletion of Zfp36l1 and Zfp36l2 in lymphocytes led to dysregulated B and T lymphopoiesis.79 In developing B cells, double knockout resulted in a loss of quiescence prior to proper assembly and expression of the pre-BCR.80 RNA sequencing and individual-nucleotide resolution cross-linking and immunoprecipitation revealed coordination of transcripts that limit cell cycle progression. Loss of both Zfp36l1 and Zfp36l2 during thymopoiesis led to initial thymic atrophy associated with a loss of cell cycle control, suggesting the existence of a conserved regulon of quiescence that is essential for correct antigen receptor gene rearrangement during lymphocyte development.81 As these double-knockout mice aged further, they developed T-cell lymphoblastic leukemia, in part as a result of deregulated stability and expression of Notch1 mRNA.79
The hypothesis that these genes might function as tumor suppressors in human malignancy was supported by a recent pan-cancer genomic analysis that identified evidence of strong selective pressure for inactivating mutations of ZFP36L1 and ZFP36L2 across multiple tissue types, suggesting a conserved mechanism of tumor control.82 Indeed, recent lymphoma sequencing studies have identified recurrent inactivating mutations of ZFP36L1.83,84 A likely hypothesis is that these mutations act to promote cell cycle progression in tumor cells; however, recent observations suggest alternative mechanisms of tumor promotion. Oncogenic RAS signaling in different cancer types led to increased expression of PD-L1 on tumor cells and, hence, suppression of the host antitumor immune response.85 This effect was mediated by RAS-induced phosphorylation of ZFP36, which abolished the ability of ZFP36 to bind and destabilize PD-L1 mRNA. Indeed, oncogenic signaling activity of MAPK and phosphatidylinositol 3-kinase pathways may lead to global inactivation of ZFP36 proteins, with downstream tumor-promoting effects on cell cycle control and immune interaction.
NMD leads to the degradation of transcripts containing premature stop codons located prior to the final exon and those with especially long 3′UTRs.
A dominant mechanism promoting NMD is the accumulation of the RNA helicase UPF1 downstream of the termination codon. RBPs, including PTBP1 and hnRNPL, are able to antagonize UPF1 accumulation by sequence-specific interaction with subsets of mRNAs. This mechanism is coopted by lymphoma cells carrying the t(14:18) translocation between BCL2 and the immunoglobulin heavy chain locus. The resulting fusion transcript retains the BCL2 stop codon, followed by several downstream immunoglobulin heavy chain exons that would normally promote NMD and abrogate expression of BCL2 protein. Instead, a CA-rich element in the proximal 3′UTR recruits hnRNPL, which protects the fusion transcript from NMD and permits expression of BCL2 protein.86
Summary
Our understanding of the multilayered regulation imposed beyond the point of transcription is increasing. Much of this regulation is mediated by RBPs, which allow coordinated remodeling of the transcriptome and proteome in response to microenvironmental stimuli. An emerging feature of many RBPs is their frequent involvement in different aspects of RNA metabolism. Thus, genetic or posttranslational changes to an individual RBP may have consequences for many RNA targets, as well as at multiple points of their RNA metabolism. An increased understanding of how RBPs influence this network of regulation in normal and malignant hematopoiesis is already revealing new strategies for therapeutic targeting. These strategies may target the signaling pathways that control RBPs, the RBP itself, the RBP–RNA interaction, or the downstream alterations to the proteome mediated by changes in RBP function.
Acknowledgments
The authors thank the Medical Research Council, The Biotechnology and Biological Sciences Research Council BBS/E/B/000C0428, Bloodwise, Cancer Research Therapeutics, and Wellcome for funding research in the authors’ laboratories, as well as Özge Glzlenci for comments on the manuscript.
Authorship
Contribution: D.J.H., M.S., and M.T. planned the outline of the manuscript; D.J.H. wrote the first draft; M.T. and M.S. revised and edited manuscript drafts; and M.S. designed and prepared the figures.
Conflict-of-interest disclosure: D.J.H. has received research funding from Gilead Sciences. M.T. has received research support from Cancer Research Technologies and consultancy fees from Roche. M.S. declares no competing financial interests.
Correspondence: Daniel J. Hodson, Wellcome–MRC Cambridge Stem Cell Institute, Cambridge Biomedical Campus, Cambridge CB2 0AH, United Kingdom; e-mail: djh1002@cam.ac.uk; and Martin Turner, Laboratory of Lymphocyte Signaling and Development, Babraham Institute, Babraham Research Campus, Cambridge CD22 3AT, United Kingdom; e-mail: martin.turner@babraham.ac.uk.
