

Key Points
The absence of Cdk6 ameliorates MPN hallmarks, lessens splenomegaly, and enhances survival of JAK2V617F mice.
CDK6 facilitates MPN by enhancing cytokine production (in conjunction with NF-κB), activating leukemic stem cells and preventing apoptosis.

Abstract
Over 80% of patients with myeloproliferative neoplasms (MPNs) harbor the acquired somatic JAK2V617F mutation. JAK inhibition is not curative and fails to induce a persistent response in most patients, illustrating the need for the development of novel therapeutic approaches. We describe a critical role for CDK6 in MPN evolution. The absence of Cdk6 ameliorates clinical symptoms and prolongs survival. The CDK6 protein interferes with 3 hallmarks of disease: besides regulating malignant stem cell quiescence, it promotes nuclear factor κB (NF-κB) signaling and contributes to cytokine production while inhibiting apoptosis. The effects are not mirrored by palbociclib, showing that the functions of CDK6 in MPN pathogenesis are largely kinase independent. Our findings thus provide a rationale for targeting CDK6 in MPN.
Introduction
Myeloproliferative neoplasms (MPNs) are clonal hematological stem cell disorders characterized by uncontrolled expansion of one or more myeloid lineages. Patients with MPN are at risk of bone marrow (BM) fibrosis and occurrence of thromboembolic events, conditions that contribute to a poor clinical outcome. A major complication of MPNs is transformation to secondary acute myeloid leukemia, which is also associated with a lowered life expectancy.
The majority of MPNs arise due to somatic mutations that result in constitutively active tyrosine kinase signaling cascades, providing the malignant cells with a gain of fitness. The first clinical insights into MPN pathogenesis stemmed from the discovery of a single gain-of-function point mutation (Val617Phe) in the nonreceptor tyrosine kinase JAK2 (JAK2V617F) in >95% of patients with polycythemia vera (PV) and in 50% to 60% of patients with essential thrombocythemia and primary myelofibrosis.1-4 There have been considerable efforts to develop JAK inhibitors: the JAK1/2 kinase inhibitor ruxolitinib is approved for use in patients with advanced primary myelofibrosis and PV who are resistant to hydroxyurea. Other drugs used to treat MPN patients include interferon-α and anagrelide. However, there is a lack of curative therapies: JAK inhibitors reduce splenomegaly and control MPN-related symptoms quite effectively but fail to eradicate the malignant clone. There is an immediate need for additional therapeutic strategies.
MPN patients are characterized by elevated levels of circulating proinflammatory cytokines arising in both malignant and nonmalignant hematopoietic cells.5 Increased cytokine production is linked to adverse prognosis and contributes to clinical symptoms, BM fibrosis, and extramedullary hematopoiesis.6 The chronic inflammatory state is mediated by molecular cross talk between 2 key regulators of inflammation: the JAK2 downstream target STAT3 and the transcription factor nuclear factor κB (NF-κB).7 NF-κB is controlled via cytosolic retention by inhibitor of NF-κB (IκB) proteins. Phosphorylation-dependent proteolytic degradation of IκBs in response to inducers such as proinflammatory cytokines (eg, interleukin [IL]-1 or tumor necrosis factor-α [TNF-α]) is followed by nuclear translocation and DNA binding of NF-κB subunits. Within the nuclear compartment, posttranslational modifications provide an additional layer of regulation of NF-κB activity. Transactivation of the transcriptionally most potent NF-κB subunit p65 is enhanced by phosphorylation at Ser536 by cyclin-dependent kinase 6 (CDK6). This activation is required for the interaction between corepressors and coactivators required for the expression of inflammatory genes.8
CDK6 and its close relative CDK4 are critical regulators of cell-cycle progression. In complex with cyclin D, CDK4 and CDK6 have redundant roles in relieving retinoblastoma-mediated transcriptional repression to promote exit from the G1 phase of the cell cycle. The alteration or inhibition of 1 of these 2 cell-cycle kinases alone does not suffice to inhibit cell-cycle progression, but the simultaneous deletion of both kinases induces late embryonic lethality in mice due to defects in hematopoiesis.9,10 Loss of Cdk6 alone is not lethal but leads to defects in hematopoietic cell proliferation and mild anemia.10,11 There is ample evidence for additional substrates and functional differences between CDK4 and CDK6 that go well beyond the control of the cell cycle. In a variety of human lymphomas and leukemias,10,12-23 the CDK6 gene is frequently amplified or overexpressed, a feature not shared by CDK4. CDK6 but not CDK4 is a direct regulator of transcription in both kinase-dependent and -independent manners, interacting with a range of transcription factors, including STATs and AP-1.11,24-27 It also serves as a nuclear cofactor for NF-κB p65 and mediates CXCL8 (IL-8) expression.25 In addition, CDK6 stabilizes the cytoskeletal integrity of erythroid cells on a transcriptional and structural level.11 The cycling of human hematopoietic stem cells (HSCs) depends on the level of CDK6,28 and under stress conditions, CDK6 contributes to quiescence of murine stem cells by the transcriptional regulation of Egr1.26
We now report a new component of the oncogenic mechanisms that underlie MPN pathobiology in JAK2V617F mice: the absence of Cdk6 attenuates clinical symptoms. In JAK2V617F progenitor cells, the CDK6 protein acts as a transcriptional regulator of NF-κB signaling, apoptosis, and HSC activation. The cell-cycle kinase CDK6 is thus required to sustain the fitness of JAK2V617F-transformed progenitors to maintain disease. A CDK6/NF-κB–dependent axis contributes to regulating the levels of circulating cytokines in JAK2V617F mutant mice. Our data thus identify CDK6 as a molecular node that integrates NF-κB–dependent inflammation, apoptosis, and malignant stem cell activation in JAK2V617F-mediated MPN and provide a rationale for therapeutic evaluation of CDK6 inhibition in this disease.
Methods
Mouse strains and transplantation studies
Mice were maintained under specific pathogen-free conditions at the Institute of Pharmacology and Toxicology, University of Veterinary Medicine, Vienna and at the Institute of Molecular Biotechnology, Vienna. Mice carrying a floxed heterozygous conditional knock-in (KI) allele of JAK2V617F 29,30 were crossed with VavCre and Cdk6−/− mice.31 All strains were bred on the C57BL/6N background. NOD/SCID/IL-2Rγ−/− (NSG) and B6.SJL-Ptprca (Ly5.1+) mice were bred at the University of Veterinary Medicine, Vienna. Eight- to 32-week-old mice were used for experiments. All procedures were approved by the institutional ethics and animal welfare committee of the University of Veterinary Medicine, Vienna (BMWFW-68.205/0112-WF/V/3b/2016, BMWF-68.205/0103-WF/V/3b/2015, BMWFW-68.205/0093-WF/V/3b/2015) and at the Institute of Molecular Biotechnology, Vienna (BMWFW-66.015/0004-WF/V/3b/2016) and the national authority according to §§26ff. of the Animal Experiment Act, Tierversuchsgesetz 2012-TVG 2012.
RNA sequencing and bioinformatics analysis
Purified Lineage−Sca1+cKit+ (LSK) cells were isolated from BM of 8-week-old VavCre; Jak2+/+; Cdk6+/+, VavCre; JAK2V617F; Cdk6+/+, VavCre; JAK2V617F; Cdk6−/−, VavCre; Jak2+/+; Cdk6−/− mice and VavCre; JAK2V617F; Cdk6+/+ mice treated with palbociclib. RNA was extracted using the RNeasy Micro Kit according to the manufacturer’s instructions (Qiagen, Venlo, The Netherlands). The amount of total RNA was quantified using the Qubit 2.0 Fluorometric Quantitation system (Life Technologies, Carlsbad, CA), and the RNA integrity number was determined using the Experion Automated Electrophoresis System (Bio-Rad, Hercules, CA). RNA-seq libraries were prepared with the TruSeq Stranded mRNA LT sample preparation kit (Illumina, San Diego, CA) using both Sciclone and Zephyr liquid handling robotics (PerkinElmer, Waltham, MA). Library concentrations were quantified with the Qubit 2.0 Fluorometric Quantitation system (Life Technologies), and the size distribution was assessed using the Experion Automated Electrophoresis System (Bio-Rad). For sequencing, samples were diluted and pooled into NGS libraries in equimolar amounts. Expression profiling libraries were sequenced on Illumina HiSeq 3000/4000 instruments in 50-base-pair single-end mode, and base calls provided by the Illumina Realtime Analysis software were subsequently converted into BAM format (Illumina2bam) before demultiplexing (BamIndexDecoder) into individual, sample-specific BAM files via Illumina2bam tools (1.17.3 https://github.com/wtsi-npg/illumina2bam). After quality control of raw data with FastQC, NGS reads were trimmed based on quality and adapter sequence content with Trimmomatic (0.36) and mapped to the GENECODE M13 genome using STAR (2.5.2b) with default parameters. FeatureCounts from the Subread package (1.5.1) was used to obtain gene counts for union gene models. Statistical analysis was conducted in the R environment32 using the DESeq2 package33 (1.18.1) for differential expression analysis, the RUVSeq package34 (0.99.1) for removing unwanted variation in the data, and the pheatmap package (1.0.8) for visualization purposes. Differentially expressed genes with adjusted P value <.05 and absolute log2-fold change >1 were considered significant. Gene set enrichment analysis (GSEA)35,36 (2.2.4) was used for gene set enrichment analysis against Hallmark Gene Set Collection.37 False discovery rate <0.25 was considered significant.
These data can be accessed under the Gene Expression Omnibus database, accession number GSE123401.
Results
Cdk6 influences the course of MPN in JAK2V617F KI mice
To investigate the role of Cdk6 in the development of MPN, we crossed VavCre; JAK2V617F KI29,30 with Cdk6−/− mice31 (supplemental Figure 1A, available on the Blood Web site). As expected, mice expressing JAK2V617F (VavCre; JAK2V617F; Cdk6+/+) had markedly elevated red blood cell numbers, an elevated hematocrit, and more hemoglobin in the peripheral blood (PB) than wild-type (WT) control mice (VavCre; Jak2+/+; Cdk6+/+ and VavCre; Jak2+/+; Cdk6−/−) over the course of 30 weeks. In line with published reports, lymphocytes, monocytes, and granulocytes were increased, and the VavCre; JAK2V617F KI mice displayed a pronounced thrombocytosis. In the absence of Cdk6, the effects of JAK2V617F on erythropoiesis decreased as the mice aged (from 24 weeks). The absence of Cdk6 was associated with a significantly lower platelet count from an early age. The effects of Cdk6 deficiency on erythropoiesis were only evident in older JAK2V617F mice, suggesting that CDK6 exerts its effects by different mechanisms in younger and older mice, and in platelets and erythrocytes (Figure 1A).

Cdk6 deletion ameliorates symptoms associated with JAK2V617Fmutation. (A) Time course of blood counts of transgenic mice (n = 6 per genotype). Results are presented as means + standard deviation (SD). Two-way analysis of variance (ANOVA) with subsequent Bonferroni posttest was used, and significance between VavCre; JAK2V617F; Cdk6+/+ and VavCre; JAK2V617F; Cdk6−/− is indicated. *P < .05; **P < .01;***P < .001; ****P < .0001. HCT, hematocrit; HGB, hemoglobin; n.s., not significant; RBC, red blood cells. (B) Survival of mice is shown as Kaplan-Meier curves. The experiment was terminated after 365 days. Mean survival, 98 days (VavCre; JAK2V617F; Cdk6+/+), 316 days (VavCre; JAK2V617F; Cdk6−/−). Log-rank test was used for statistical comparison. *P < .05; ****P < .0001. The group sizes were n (VavCre; JAK2V617F; Cdk6+/+) = 23, n (VavCre; JAK2V617F; Cdk6−/−) = 23, n (VavCre; Jak2+/+; Cdk6+/+) = 10, and n (VavCre; Jak2+/+; Cdk6−/−) = 10. (C) Spleen weights at 8 weeks of age are depicted. The group sizes were n (VavCre; Jak2+/+; Cdk6+/+) = 19, n (VavCre; JAK2V617F; Cdk6+/+) = 15, n (VavCre; JAK2V617F; Cdk6−/−) = 13, and n (VavCre; Jak2+/+; Cdk6−/−) = 15. Error bars indicate + SD. One-way ANOVA with subsequent Bonferroni posttest was used, and significance is indicated. **P < .01; ****P < .0001.
Cdk6 deletion ameliorates symptoms associated with JAK2V617Fmutation. (A) Time course of blood counts of transgenic mice (n = 6 per genotype). Results are presented as means + standard deviation (SD). Two-way analysis of variance (ANOVA) with subsequent Bonferroni posttest was used, and significance between VavCre; JAK2V617F; Cdk6+/+ and VavCre; JAK2V617F; Cdk6−/− is indicated. *P < .05; **P < .01;***P < .001; ****P < .0001. HCT, hematocrit; HGB, hemoglobin; n.s., not significant; RBC, red blood cells. (B) Survival of mice is shown as Kaplan-Meier curves. The experiment was terminated after 365 days. Mean survival, 98 days (VavCre; JAK2V617F; Cdk6+/+), 316 days (VavCre; JAK2V617F; Cdk6−/−). Log-rank test was used for statistical comparison. *P < .05; ****P < .0001. The group sizes were n (VavCre; JAK2V617F; Cdk6+/+) = 23, n (VavCre; JAK2V617F; Cdk6−/−) = 23, n (VavCre; Jak2+/+; Cdk6+/+) = 10, and n (VavCre; Jak2+/+; Cdk6−/−) = 10. (C) Spleen weights at 8 weeks of age are depicted. The group sizes were n (VavCre; Jak2+/+; Cdk6+/+) = 19, n (VavCre; JAK2V617F; Cdk6+/+) = 15, n (VavCre; JAK2V617F; Cdk6−/−) = 13, and n (VavCre; Jak2+/+; Cdk6−/−) = 15. Error bars indicate + SD. One-way ANOVA with subsequent Bonferroni posttest was used, and significance is indicated. **P < .01; ****P < .0001.
As observed by others, heterozygous JAK2V617F (VavCre; JAK2V617F; Cdk6+/+) animals develop a lethal MPN with 100% penetrance, and their median survival was 98 days. In contrast, the VavCre; JAK2V617F; Cdk6−/− cohort has a median survival of 316 days. All Jak2+/+ animals with or without Cdk6 remained disease free (Figure 1B).
The MPN phenotype observed in JAK2V617F transgenic mice is associated with prominent splenomegaly that increases with age. The average spleen weight in 8-month-old VavCre; JAK2V617F; Cdk6+/+ animals is 15-fold higher than in age-matched WT controls. The absence of Cdk6 reduces the severity of splenomegaly in JAK2V617F mutant mice; spleen weights are only marginally enhanced (1.5- or threefold) in 2- and 8-month-old mice (Figure 1C; supplemental Figure 1B-D). This indicates that Cdk6 accelerates the myeloproliferative neoplastic progression in JAK2V617F mutant mice.
Histopathological evaluation of BM and spleen sections from JAK2V617F KI mice reveals prominent megakaryocytosis and megakaryocyte dyspoiesis featuring micromegakaryocytes, macromegakaryocytes (frequently with foamy vacuolated cytoplasm), megakaryocytes with variable nuclear lobation (from hypolobated to hypersegmented), and megakaryocytes with multiple nuclei (Figure 2A; supplemental Figure 2A-F). The sections also reveal an increased proportion of myeloid cells among the erythroid precursors, although the extents differ. Pronounced myeloid proliferation reminiscent of myeloid leukemia is evident in some JAK2V617F KI mice. Lymphoid follicles are frequently obscured by the myeloproliferative neoplastic process in spleens of JAK2V617F KI mice, whereas follicular architecture is normal in the absence of Cdk6 (supplemental Figure 2E). These features are not evident in WT Jak2 mice with or without Cdk6 (Figure 2A; supplemental Figure 2A-D). Equal numbers of megakaryocytes are found in the BM of 2-month-old mice (Figure 2B). Flow cytometry profiling38 revealed enhanced numbers of late erythroid precursors in young JAK2V617F mutant mice when Cdk6 is absent (Figure 2C-E).
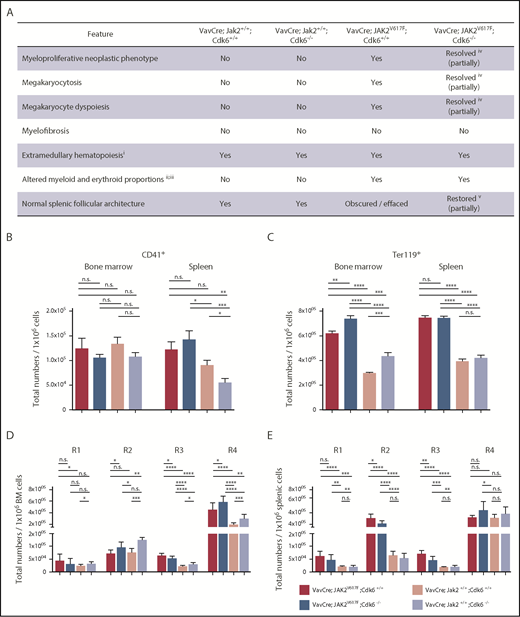
Histopathology and analysis of erythroid precursors in Cdk6-deficient JAK2V617Ftransgenic mice. Mice were euthanized at 8 weeks of age. (A) Summary of histopathologic features of 8-week-old mice with indicated genotypes. (i) Light microscopically discernible presence of erythroid and myeloid cells in the splenic red pulp indicative of an active extramedullary hematopoietic process. (ii) Nonquantitative assessment of the histologic presence of erythroid and myeloid cells. (iii) Variably increased representation of myeloid cells. (iv) Variable degree of resolution of the neoplastic process and features of dyspoiesis (modest in some samples and much more in some others). (v) Variable restoration of splenic follicular architecture (marginal to modest in some samples and much more in some others). Total numbers of CD41+ megakaryocytes (B) or total numbers of Ter119+ erythrocytes (C) per 1 × 106 cells are shown for BM and spleen. Error bars indicate + SD *P < .05; **P < .01; ***P < .001; ****P < .0001. (D-E) Total numbers of erythroid precursors per 1 × 106 cells are shown for BM (D) and spleen (E). Ter119medCD71high (proerythroblasts), Ter119highCD71high (basophilic erythroblasts), Ter119highCD71med (late basophilic and polychromatophilic erythroblasts), and Ter119highCD71low (orthochromatophilic erythroblasts) represent the regions R1-R4, respectively. Statistical analysis was performed with one-way ANOVA with subsequent Bonferroni posttest, and significance is indicated (n ≥ 10 per genotype). *P < .05; **P < .01; ***P < .001; ****P < .0001.
Histopathology and analysis of erythroid precursors in Cdk6-deficient JAK2V617Ftransgenic mice. Mice were euthanized at 8 weeks of age. (A) Summary of histopathologic features of 8-week-old mice with indicated genotypes. (i) Light microscopically discernible presence of erythroid and myeloid cells in the splenic red pulp indicative of an active extramedullary hematopoietic process. (ii) Nonquantitative assessment of the histologic presence of erythroid and myeloid cells. (iii) Variably increased representation of myeloid cells. (iv) Variable degree of resolution of the neoplastic process and features of dyspoiesis (modest in some samples and much more in some others). (v) Variable restoration of splenic follicular architecture (marginal to modest in some samples and much more in some others). Total numbers of CD41+ megakaryocytes (B) or total numbers of Ter119+ erythrocytes (C) per 1 × 106 cells are shown for BM and spleen. Error bars indicate + SD *P < .05; **P < .01; ***P < .001; ****P < .0001. (D-E) Total numbers of erythroid precursors per 1 × 106 cells are shown for BM (D) and spleen (E). Ter119medCD71high (proerythroblasts), Ter119highCD71high (basophilic erythroblasts), Ter119highCD71med (late basophilic and polychromatophilic erythroblasts), and Ter119highCD71low (orthochromatophilic erythroblasts) represent the regions R1-R4, respectively. Statistical analysis was performed with one-way ANOVA with subsequent Bonferroni posttest, and significance is indicated (n ≥ 10 per genotype). *P < .05; **P < .01; ***P < .001; ****P < .0001.
JAK2V617F-transformed HSCs and progenitor cells depend on Cdk6 for their fitness
The MPN-initiating cell is in the HSC compartment,39 where the disease originates, with the JAK2V617F mutation detectable in HSCs of human MPN patients.4 Because Cdk6 plays a key part in activating stem cells under stress,26 we investigated the effects of Cdk6 on HSC number and function in the presence of the JAK2V617F mutation, which will induce oncogenic and/or replicative stress. LSK cells in BM of young JAK2V617F mice (2 months old) are increased, and the proportion is further enhanced in the absence of Cdk6, although the overall number of cells in the BM is nearly unaltered (Figure 3A,E; supplemental Figure 3A). We attribute the change in LSK numbers to the accumulation of CD150+CD48−LSKs (long-term [LT]-HSC) and CD150+CD48+LSKs (short-term [ST]-HSC)40 in the VavCre; JAK2V617F; Cdk6−/− cohort. In accordance with our previous publication,26 the LT-HSC fraction in JAK2V617F BM was enriched with the most dormant HSCs (CD150+CD48+CD135−CD34− LSK) when Cdk6 was absent (supplemental Figure 3B). The proportion of CD150−CD48+LSKs (MPP) cells40 remained largely unchanged (Figure 3B,F). Similar alterations were found in the spleens of 2-month-old VavCre; JAK2V617F; Cdk6−/− mice (supplemental Figure 3C-D,G).
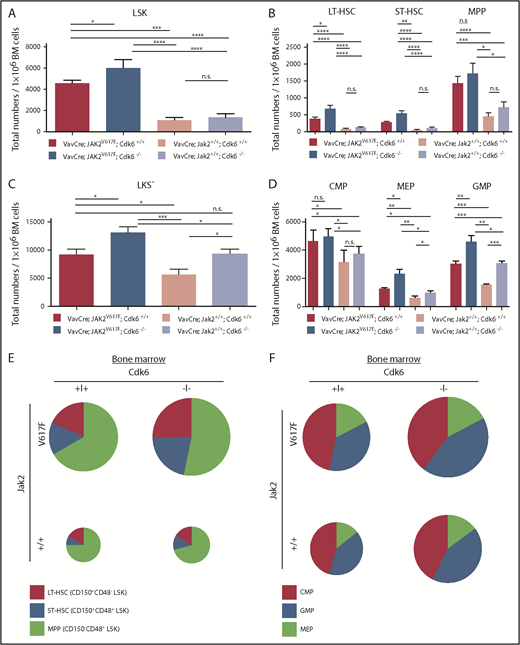
Altered composition of the hematopoietic stem and progenitor cells upon Cdk6 ablation in JAK2V617Fmutant BM. Mice were euthanized at 8 weeks of age. (A-D) Total numbers of HSCs and progenitors per 1 × 106 BM cells are shown. Error bars indicate + SD. *P < .05; **P < .01; ***P < .001; ****P < .0001. The group sizes were n ≥ 10 per genotype. CMP (common myeloid progenitor), LKS−CD34intCD16/32int; GMP (granulocyte/macrophage progenitor): LKS−CD34+CD16/32+; LT-HSC, CD150+CD48−LSK; MEP (megakaryocyte/erythroid progenitor): LKS−CD34−CD16/32−. (E-F) Overview of distribution of HSCs (E) and myeloid progenitors (F) in BM. Chart size corresponds to absolute numbers of LSK (E) and LKS− (F) cells in mice of respective genotype.
Altered composition of the hematopoietic stem and progenitor cells upon Cdk6 ablation in JAK2V617Fmutant BM. Mice were euthanized at 8 weeks of age. (A-D) Total numbers of HSCs and progenitors per 1 × 106 BM cells are shown. Error bars indicate + SD. *P < .05; **P < .01; ***P < .001; ****P < .0001. The group sizes were n ≥ 10 per genotype. CMP (common myeloid progenitor), LKS−CD34intCD16/32int; GMP (granulocyte/macrophage progenitor): LKS−CD34+CD16/32+; LT-HSC, CD150+CD48−LSK; MEP (megakaryocyte/erythroid progenitor): LKS−CD34−CD16/32−. (E-F) Overview of distribution of HSCs (E) and myeloid progenitors (F) in BM. Chart size corresponds to absolute numbers of LSK (E) and LKS− (F) cells in mice of respective genotype.
Because JAK2V617F-positive disorders originate from an early stem cell with multipotent potential (able to differentiate to the myeloid and lymphoid lineages),41 we examined the myeloid progenitor compartment. Young adult VavCre; JAK2V617F; Cdk6−/− mice accumulate myeloid-primed progenitors (LKS−) due to increased proportions of megakaryocyte/erythroid progenitor and in the BM but not in the spleen (Figure 3C-D,F; supplemental Figure 3E-F,H). Nevertheless, the proportion of Gr1+/Mac1+ cells is drastically reduced in the BM (and the spleen) of JAK2V617F mice without Cdk6 (Figure 4A-C). Similarly, VavCre; JAK2V617F; Cdk6−/− animals harbor decreased numbers of monocytes, eosinophils, and neutrophils (in PB only by trend), as confirmed by colony formation assays (Figure 4D; supplemental Figures 4 and 5). The influence of Cdk6 is not restricted to the myeloid compartment: the lymphoid compartment also has significantly different B- and T-cell fractions in VavCre; JAK2V617F; Cdk6−/− mice (Figure 4A-C). The findings are depicted graphically in Figure 3E-F and supplemental Figure 3F-G, in which the size of the circles indicates the total number of cells in the BM or spleen. The results are consistent with the idea that Cdk6 has tissue- and stage-specific functions in regulating lymphoid and myeloid differentiation.
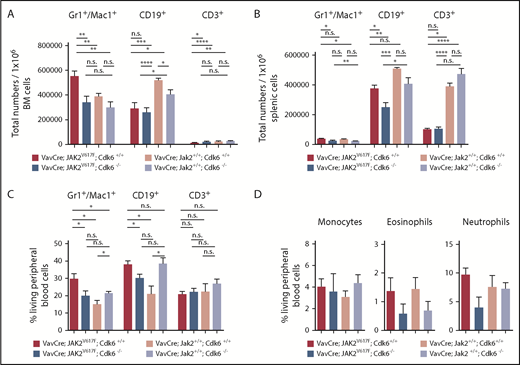
Altered composition of the myeloid lineages upon Cdk6 ablation in JAK2V617Fmutant mice. Mice were euthanized at 8 weeks of age (n ≥ 10 per genotype). (A-B) Total numbers of indicated lineages per 1 × 106 BM (A) and splenic cells (B) are shown. Error bars indicate + SD * P < .05; **P < .01; ****P < .0001. (C) Frequencies of indicated lineages in PB cells from mice used in panels A and B are depicted. Results are presented as means + SD *P < .05. (D) Percentage of indicated myeloid populations in PB is shown. Error bars indicate + standard error of the mean. n ≥ 3 per genotype; monocytes MHCII−CD11b+CD115+Ly6C−; neutrophils CD11b+Ly6G+Ly6ClowCD11c−CD115−CD170−; eosinophils CD11b+Ly6G−Ly6CmidCD11c−.
Altered composition of the myeloid lineages upon Cdk6 ablation in JAK2V617Fmutant mice. Mice were euthanized at 8 weeks of age (n ≥ 10 per genotype). (A-B) Total numbers of indicated lineages per 1 × 106 BM (A) and splenic cells (B) are shown. Error bars indicate + SD * P < .05; **P < .01; ****P < .0001. (C) Frequencies of indicated lineages in PB cells from mice used in panels A and B are depicted. Results are presented as means + SD *P < .05. (D) Percentage of indicated myeloid populations in PB is shown. Error bars indicate + standard error of the mean. n ≥ 3 per genotype; monocytes MHCII−CD11b+CD115+Ly6C−; neutrophils CD11b+Ly6G+Ly6ClowCD11c−CD115−CD170−; eosinophils CD11b+Ly6G−Ly6CmidCD11c−.
The numbers of cells in the BM of 8-month-old VavCre; JAK2V617F; Cdk6−/− mice were markedly lower than those of their JAK2V617F counterparts (supplemental Figure 6A). There were proportionally more LSKs, including LT-HSCs and ST-HSCs, in the BM and spleen of 32-week-old JAK2V617F mutant mice lacking Cdk6, and there appeared to be more myeloid precursors (LKS−) in the BM and spleen (supplemental Figure 6B-C). As in young animals, the absence of Cdk6 alters the lymphoid/myeloid lineage (supplemental Figure 6D-F), consistent with a role of CDK6 in the effects of JAK2V617F on the hematopoietic pool.
To test whether the effects of Cdk6 on JAK2V617F-induced MPN are cell autonomous, we transplanted total BM cells from VavCre; JAK2V617F; Cdk6+/+ or VavCre; JAK2V617F; Cdk6−/− transgenic mice into nonirradiated NSG mice. The procedure triggers disease that depends on the constant replenishment of peripheral cells by JAK2V617F-positive malignant stem cells. Recipient mice were sacrificed upon signs of clinical illness. Similar to primary Cdk6-deficient JAK2V617F mice, recipients of the BM cells from VavCre; JAK2V617F; Cdk6−/− mice exhibit extended disease latency and have a significant survival benefit. Mice that receive Cdk6-deficient BM cells without a Jak2 mutation (VavCre; Jak2+/+; Cdk6−/−) remain disease free (Figure 5A). This confirms that CDK6 has effects within the hematopoietic cell compartment. Megakaryocytic abnormalities and myeloid and megakaryocytic proliferation are less pronounced in recipients of VavCre; JAK2V617F; Cdk6−/− BM than in diseased animals transplanted with VavCre; JAK2V617F; Cdk6+/+ BM (Figure 5B; supplemental Figure 7A), although there is no significant difference in spleen weight (supplemental Figure 7B). Increased numbers of LT-HSCs in the spleen (Figure 5C; supplemental Figure 7C) are consistent with the reduced capacity of Cdk6-deficient JAK2V617F mutant progenitors to replenish the blood system, although there is no effect on myeloid precursors (LKS−) (Figure 5D; supplemental Figure 7D). The effects cannot be explained by an altered homing capacity of Cdk6-deficient stem cells (Figure 5E).

Cdk6 provides JAK2V617Fmutant progenitors with a gain of fitness to maintain disease. Mice were euthanized when terminally ill. (A) Kaplan-Meier plot depicting disease onset of immune-compromised NSG recipients injected with 1 × 106 total BM cells of indicated genotype. Mean survival, 180 days (VavCre; JAK2V617F; Cdk6+/+, n = 13); mean survival, 267.5 days (VavCre; JAK2V617F; Cdk6−/−, n = 8). Recipients transplanted with WT Jak2 BM with or without Cdk6 (VavCre; Jak2+/+; Cdk6+/+ and VavCre; Jak2+/+; Cdk6−/−) did not disease. Log-rank test was used for statistical comparison. n = 4 per genotype; **P < .01; ***P < .001; ****P < .0001. (B) Hematoxylin-eosin staining of BM (scale bars, 100 μm; higher magnifications, 50 μm) and spleen (scale bars, 200 μm; higher magnifications, 50 μm). (C-D) Fold change of frequencies of HSCs (C) and myeloid progenitors (D) in BM and spleen samples of recipients transplanted with VavCre; JAK2V617F; Cdk6−/− BM cells (VavCre; JAK2V617F; Cdk6+/+ controls set to 1; ***P < .001). (E) Homing in % of Ly5.2+ LSKs in 2.5 × 106 BM cells of Ly5.1+ recipient mice.
Cdk6 provides JAK2V617Fmutant progenitors with a gain of fitness to maintain disease. Mice were euthanized when terminally ill. (A) Kaplan-Meier plot depicting disease onset of immune-compromised NSG recipients injected with 1 × 106 total BM cells of indicated genotype. Mean survival, 180 days (VavCre; JAK2V617F; Cdk6+/+, n = 13); mean survival, 267.5 days (VavCre; JAK2V617F; Cdk6−/−, n = 8). Recipients transplanted with WT Jak2 BM with or without Cdk6 (VavCre; Jak2+/+; Cdk6+/+ and VavCre; Jak2+/+; Cdk6−/−) did not disease. Log-rank test was used for statistical comparison. n = 4 per genotype; **P < .01; ***P < .001; ****P < .0001. (B) Hematoxylin-eosin staining of BM (scale bars, 100 μm; higher magnifications, 50 μm) and spleen (scale bars, 200 μm; higher magnifications, 50 μm). (C-D) Fold change of frequencies of HSCs (C) and myeloid progenitors (D) in BM and spleen samples of recipients transplanted with VavCre; JAK2V617F; Cdk6−/− BM cells (VavCre; JAK2V617F; Cdk6+/+ controls set to 1; ***P < .001). (E) Homing in % of Ly5.2+ LSKs in 2.5 × 106 BM cells of Ly5.1+ recipient mice.
The data are consistent with a model in which the effects of Cdk6 on JAK2V617F-induced MPN are cell autonomous and Cdk6 is required for the JAK2V617F mutation to maintain disease.
CDK6 coordinates NF-κB signaling, apoptosis and HSC activation in JAK2V617F LSKs
To investigate how Cdk6 modulates JAK2V617F-induced MPN, we compared the messenger RNA (mRNA) profiles of hematopoietic cells with or without mutations in Jak2 and Cdk6 by RNA sequencing using BM-derived LSK cells from 2-month-old mice. Principal component analysis identified WT Jak2 vs JAK2V617F as the dominant factor (principal component 1) that explains 50% of the variation (supplemental Figure 8A). The second most prominent difference was attributed to loss of Cdk6 vs WT Cdk6, which accounts for 17% of the variation. Cdk6 ablation in the context of JAK2V617F led to altered levels of >60 protein-coding transcripts (Figure 6A; supplemental Table 1; supplemental Figure 8B). We overlaid the list of altered transcripts with gene sets for bipotent megakaryocyte/erythroid progenitors, granulocyte/macrophage progenitors, and common lymphoid progenitors.42 The enrichment of megakaryocyte/erythroid progenitors and granulocyte/macrophage progenitors genes confirmed the early myeloid bias of JAK2V617F LSKs in the absence of Cdk6 (Figure 3D; supplemental Figure 8C). In Cdk6-deficient JAK2V617F mutant LSKs, competitive GSEA uncovered an enrichment of apoptosis signatures and TNF-α signaling via NF-κB, which contained predominantly negative regulators of NF-κB signaling (Figure 6B; supplemental Figure 8D).
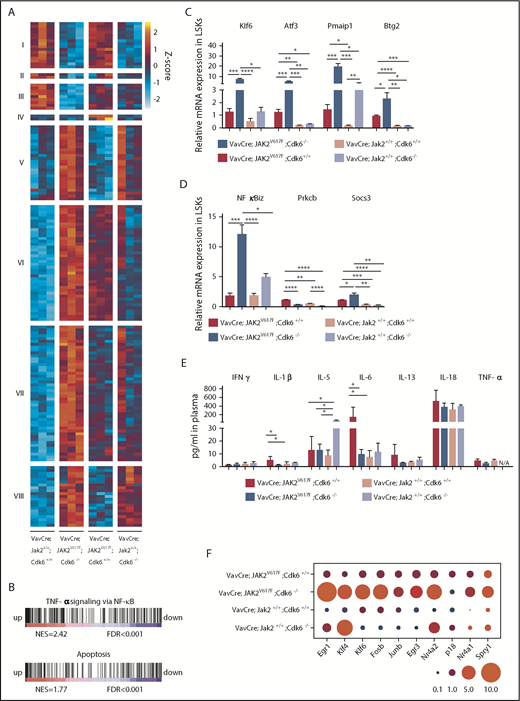
CDK6 acts as a central node in apoptotic network, NF-κB signaling, and HSC activation in JAK2V617Fmutant LSKs. (A) Heat map shows differentially expressed genes from LSKs of indicated genotype (adjusted P ≤ .05; absolute log2-fold changes >1; contrast: VavCre; JAK2V617F; Cdk6−/− vs VavCre; Jak2+/+; Cdk6+/+). Regularized log-transformation of count data was used as input for the heat map. For full data set, see supplemental Figure 9B and supplemental Table 1. Color code: red, upregulation; blue, downregulation. n = 3 per genotype. (B) Gene set enrichment analysis for gene expression signatures of apoptosis and TNF-α signaling via NF-κB in LSKs of VavCre; JAK2V617F; Cdk6−/− compared with VavCre; JAK2V617F; Cdk6+/+. For full data set, see supplemental Figure 8D. Color code: red, upregulation; blue, downregulation. FDR, false discovery rate; NES, normalized enrichment score. (C-D) Expression of indicated genes was determined by quantitative real time polymerase chain reaction in LSKs of respective genotype. Relative expression levels were normalized to the housekeeping genes Rplp0 and Hprt. Results are presented as means + SD (n = 3 per genotype). One-way ANOVA with subsequent Bonferroni posttest was used, and significance is indicated. *P < .05; **P < .01; ***P < .001; ****P < .0001. (E) Levels of indicated proinflammatory cytokines were measured in plasma samples of respective genotype. Error bars indicate + SD (n ≥ 5 per genotype). One-way ANOVA with subsequent Bonferroni posttest was used, and significance is indicated *P < .05. IFN, interferon; N/A, not detected. (F) Bubble plot showing relative mRNA levels for indicated genes determined by quantitative real time polymerase chain reaction in LSKs (n = 3 per genotype). Relative expression levels were normalized to the housekeeping genes Rplp0 and Hprt. Circled area corresponds to relative expression. Significance is indicated in supplemental Figure 9C.
CDK6 acts as a central node in apoptotic network, NF-κB signaling, and HSC activation in JAK2V617Fmutant LSKs. (A) Heat map shows differentially expressed genes from LSKs of indicated genotype (adjusted P ≤ .05; absolute log2-fold changes >1; contrast: VavCre; JAK2V617F; Cdk6−/− vs VavCre; Jak2+/+; Cdk6+/+). Regularized log-transformation of count data was used as input for the heat map. For full data set, see supplemental Figure 9B and supplemental Table 1. Color code: red, upregulation; blue, downregulation. n = 3 per genotype. (B) Gene set enrichment analysis for gene expression signatures of apoptosis and TNF-α signaling via NF-κB in LSKs of VavCre; JAK2V617F; Cdk6−/− compared with VavCre; JAK2V617F; Cdk6+/+. For full data set, see supplemental Figure 8D. Color code: red, upregulation; blue, downregulation. FDR, false discovery rate; NES, normalized enrichment score. (C-D) Expression of indicated genes was determined by quantitative real time polymerase chain reaction in LSKs of respective genotype. Relative expression levels were normalized to the housekeeping genes Rplp0 and Hprt. Results are presented as means + SD (n = 3 per genotype). One-way ANOVA with subsequent Bonferroni posttest was used, and significance is indicated. *P < .05; **P < .01; ***P < .001; ****P < .0001. (E) Levels of indicated proinflammatory cytokines were measured in plasma samples of respective genotype. Error bars indicate + SD (n ≥ 5 per genotype). One-way ANOVA with subsequent Bonferroni posttest was used, and significance is indicated *P < .05. IFN, interferon; N/A, not detected. (F) Bubble plot showing relative mRNA levels for indicated genes determined by quantitative real time polymerase chain reaction in LSKs (n = 3 per genotype). Relative expression levels were normalized to the housekeeping genes Rplp0 and Hprt. Circled area corresponds to relative expression. Significance is indicated in supplemental Figure 9C.
The significant dysregulation of a set of cell-death regulators in Cdk6-deficient JAK2V617F LSKs, including Btg2,43 Pmaip1 (Noxa),44 Klf6,45,46 and its downstream target Atf3,43,46,47 was verified by quantitative polymerase chain reaction (Figure 6C). We also observed a Cdk6-dependent alteration in the expression of genes regulating NF-κB signaling: Cdk6-deficient LSK cells displayed a pronounced upregulation of NF-κB inhibitor zeta (NFκBiz) and of suppressor of cytokine signaling 3 (Socs3), a negative regulator of JAK/STAT activation and NF-κB signaling,48-51 as well as a downregulation of Prkcb, a protein kinase involved in activating NF-κB activation and frequently overexpressed in JAK2V617F+ PV patients52 (Figure 6D). Cdk6 is thus involved in regulating the levels of circulating inflammatory cytokines linked to the onset of JAK2V617F-positive MPN. A Luminex-based profiling is consistent with this conclusion: the levels of IL-6 and IL-1β, 2 cytokines essential for myeloid lineage output, are significantly decreased in JAK2V617F plasma upon Cdk6 deletion (Figure 6E). These alterations presumably contribute to impaired myeloproliferation and the reduced myeloid expansion.
The core genes in the GSEA signal “TNF-α signaling via NF-κB” (supplemental Figure 9A) include many genes known to regulate stem cells. Because CDK6 has recently been shown to represent a transcriptional node that controls stem cell quiescence under stress conditions,26 we examined the signature genes associated with CDK6 on a JAK2V617F background, looking for genes that regulate quiescence and proliferation of HSCs (supplemental Figure 9B). We found a number of mouse and human transcription factors that are known to be regulated by CDK653-65 (Figure 6F; supplemental Figures 9C and 10), which binds to their chromatin.25,66 This gene set includes the quiescence-inducer Egr126 (supplemental Table 2). When serially passaging BM cells in methylcellulose containing myeloid/erythroid-promoting cytokines (IL-3, IL-6, stem cell factor, and erythropoietin), we noticed that VavCre; JAK2V617F; Cdk6−/−–derived BM cells have a decreased replating capacity (Figure 7A; supplemental Figure 11A-B). This is consistent with the accumulation of dormant HSCs and the elevated expression of inducers of quiescence. The higher replating capacity of VavCre; JAK2V617F; Cdk6+/+ BM is not only a consequence of increased LT-HSC proliferation (supplemental Figure 11C) but also associated with a greater accumulation of annexin V+ apoptotic cells in Cdk6-deficient JAK2V617F LSKs, but not in total BM (Figure 7B; supplemental Figure 11D-E).
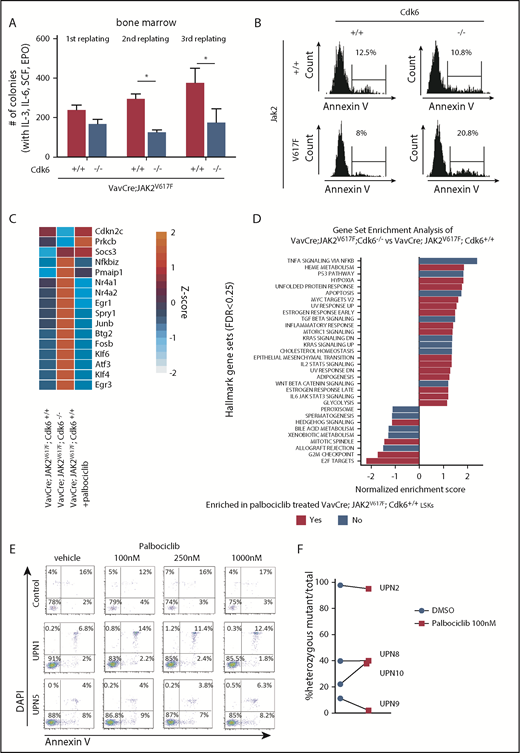
CDK6 kinase activity is not required to regulate genes involved in apoptosis and HSC quiescence in JAK2V617Fmutant LSKs. (A) Methylcellulose replating assay of whole BM cells (VavCre; JAK2V617F; Cdk6−/−, VavCre; JAK2V617F; Cdk6+/+, VavCre; Jak2+/+; Cdk6−/−, and VavCre; Jak2+/+; Cdk6+/+), 2/7 VavCre; JAK2V617F; Cdk6−/− could not be used for third replating experiments due to too small cell number. Error bars indicate + standard error of the mean (n ≥ 6 per genotype). Two-way ANOVA with Bonferroni posttest was used, and significance is indicated. *P < .05. EPO, erythropoietin; SCF, stem cell factor. (B) Representative histograms showing annexin V positivity of LSK cells in BM of indicated genotype. (C) Heat map depicts expression of validated signature genes involved in apoptosis, NF-κB signaling, and stem cell quiescence in LSKs. Regularized log-transformation of count data was used as input for the heat map. Color code: red, upregulation; blue, downregulation. Mean of 3 replicates is depicted. For full data set, see supplemental Figure 13A. (D) Overview of gene set enrichment analysis in indicated contrasts. The Hallmark Gene Set Collection was used as reference. False discovery rate <0.25 was considered significant. no: gene sets only enriched in VavCre; JAK2V617F; Cdk6−/− vs VavCre; JAK2V617F; Cdk6+/+; Yes, gene sets enriched in both VavCre; JAK2V617F; Cdk6−/− vs VavCre; JAK2V617F; Cdk6+/+ and VavCre; JAK2V617F; Cdk6+/+ palbociclib vs untreated. (E) Primary mononuclear cells were exposed to increasing concentrations of palbociclib or dimethyl sulfoxide (DMSO) for 48 hours. Apoptosis was evaluated by labeling primary CD34+CD38−CD45dim stem/progenitor cells with annexin V and 4′,6-diamidino-2-phenylindole (DAPI) followed by flow cytometry analysis. (F) Primary patient samples (1.2 × 105) were embedded in methocult with or without palbociclib. Colonies were picked at days 13 to 15, and allele-specific polymerase chain reaction was performed. Allelic burden is presented as % JAK2V617F+/− colonies vs total colony number DMSO control or palbociclib treated. DN, down; TGF, transforming growth factor; TNFA, tumor necrosis factor-α.
CDK6 kinase activity is not required to regulate genes involved in apoptosis and HSC quiescence in JAK2V617Fmutant LSKs. (A) Methylcellulose replating assay of whole BM cells (VavCre; JAK2V617F; Cdk6−/−, VavCre; JAK2V617F; Cdk6+/+, VavCre; Jak2+/+; Cdk6−/−, and VavCre; Jak2+/+; Cdk6+/+), 2/7 VavCre; JAK2V617F; Cdk6−/− could not be used for third replating experiments due to too small cell number. Error bars indicate + standard error of the mean (n ≥ 6 per genotype). Two-way ANOVA with Bonferroni posttest was used, and significance is indicated. *P < .05. EPO, erythropoietin; SCF, stem cell factor. (B) Representative histograms showing annexin V positivity of LSK cells in BM of indicated genotype. (C) Heat map depicts expression of validated signature genes involved in apoptosis, NF-κB signaling, and stem cell quiescence in LSKs. Regularized log-transformation of count data was used as input for the heat map. Color code: red, upregulation; blue, downregulation. Mean of 3 replicates is depicted. For full data set, see supplemental Figure 13A. (D) Overview of gene set enrichment analysis in indicated contrasts. The Hallmark Gene Set Collection was used as reference. False discovery rate <0.25 was considered significant. no: gene sets only enriched in VavCre; JAK2V617F; Cdk6−/− vs VavCre; JAK2V617F; Cdk6+/+; Yes, gene sets enriched in both VavCre; JAK2V617F; Cdk6−/− vs VavCre; JAK2V617F; Cdk6+/+ and VavCre; JAK2V617F; Cdk6+/+ palbociclib vs untreated. (E) Primary mononuclear cells were exposed to increasing concentrations of palbociclib or dimethyl sulfoxide (DMSO) for 48 hours. Apoptosis was evaluated by labeling primary CD34+CD38−CD45dim stem/progenitor cells with annexin V and 4′,6-diamidino-2-phenylindole (DAPI) followed by flow cytometry analysis. (F) Primary patient samples (1.2 × 105) were embedded in methocult with or without palbociclib. Colonies were picked at days 13 to 15, and allele-specific polymerase chain reaction was performed. Allelic burden is presented as % JAK2V617F+/− colonies vs total colony number DMSO control or palbociclib treated. DN, down; TGF, transforming growth factor; TNFA, tumor necrosis factor-α.
The CDK6-dependent alterations were confirmed in the human JAK2V617F HEL cell line. Knockdown of CDK6 enhanced apoptosis, as evidenced by increased poly(ADP-ribose)polymerase cleavage and elevated levels of the cell-death inducer BTG2 (supplemental Figure 12A-C). Moreover, CDK6 knockdown increases the levels of quiescence regulators and NFκBiz (supplemental Figure 12C). Exposure to IMD-0354, a potent inhibitor of NF-κB,67 induces apoptosis more potently upon CDK6 knockdown (supplemental Figure 12D).
The effects of CDK6 in JAK2V617F LSKs do not depend on its kinase activity
The CDK6 kinase is known to regulate the transcription of a number of genes and to have functions that are either dependent on or independent of its kinase activity. To evaluate the contribution of the CDK6 kinase activity to JAK2V617F-induced MPN, we examined the effects on mRNA expression profiles of LSKs from JAK2V617F KI mice treated with the CDK4/6 kinase inhibitor palbociclib (trade name IBRANCE; Pfizer) (supplemental Figure 13A). CDK6 kinase inhibition led to the dysregulation of >20 protein-coding transcripts, but apart from Socs3, the signature genes implicated in NF-κB activation, regulation of cell death, and activation of HSCs were not affected (Figure 7C; supplemental Figure 13B). Similarly, the GSEA signals “TNF-α signaling via NF-κB” and “apoptosis signatures” were not altered by palbociclib-treated JAK2V617F LSKs (Figure 7D; supplemental Figure 13C). The results show that the effects of CDK6 in LSKs are largely independent of its kinase activity. Our conclusion is supported by studies on primary mononuclear cells from the BM of JAK2V617F-positive MPN patients. Although palbociclib blocked cell-cycle progression of the bulk of MPN cells, treatment does not induce apoptosis of disease-initiating JAK2V617F-positive CD34+CD38−CD45dim stem/progenitor cells (Figure 7E; supplemental Figure 14) and has no consistent effect on allelic burden (Figure 7F). The antiapoptotic function of CDK6 in the hematopoietic cell compartment of human MPN patients is thus at least predominantly kinase independent.
Discussion
MPNs are stem cell–derived clonal myeloid malignancies with an unsatisfactory outcome. Following the discovery of dysregulated JAK/STAT signaling in patients with MPN, significant efforts have been directed toward the development of molecularly targeted therapies. The dual JAK1/2 kinase inhibitor ruxolitinib has been approved for the treatment of patients with intermediate- or high-risk myelofibrosis and PV who are resistant to hydroxyurea. Despite substantial benefits to patients, JAK inhibitors are not curative and usually have little if any effect on the mutational burden of MPN patients.
We have examined the contribution of CDK6 to the development of MPN using conditional JAK2V617F KI and Cdk6-deficient mice. We show that CDK6 has a crucial function in JAK2V617F-induced MPN progression and maintenance: the absence of Cdk6 results in a significant and lasting reduction of splenomegaly, improves disease-related symptoms, and delays persistence associated with the JAK2V617F mutation. The phenotype is reproduced by BM transplantation and is associated with changes in gene expression within the LSK compartment. CDK6 is a central signaling node that connects cell-cycle control, NF-κB–dependent inflammation, apoptosis, and malignant stem-cell function.
HSC homeostasis requires the tight regulation of cell proliferation and self-renewal to ensure the maintenance of repopulation capacity. CDK6 contributes to JAK2V617F-positive MPN by regulating the number and function of HSCs. JAK2V617F-driven stem-cell homeostasis requires Cdk6; in the absence of Cdk6, there is an increased proportion of LSK/SLAM cells, which contains the most quiescent HSC fractions, and a reduced number of peripheral cells. This indicates a road block and is parallel to the case of stress-induced hematopoiesis. CDK6 is required for stem cell activation, cell-cycle entry, and self-renewal under conditions of stress.26,28 Although CDK6 is of minor importance under steady-state conditions, under situations of stress, the suppression of the quiescence-inducer Egr1 by CDK6 is necessary for murine dormant HSCs to exit G0.26 Our data extend these findings to JAK2V617F malignant stem cells and identify additional transcriptional targets of Cdk6 in this setting. The transcriptional network regulated by CDK6 enables JAK2V617F hematopoietic stem and progenitor cells to maintain disease efficiently and to avoid apoptosis.
In our study, we failed to detect differences in the cell cycle of LSKs among the genotypes; Chen et al68 found that JAK2V617F LSKs contain more proliferating cells, whereas Mullally et al69 showed an unaltered cell-cycle distribution of JAK2V617F LSKs. These discrepancies may be explained by the use of different JAK2V617F KI models or of assays differing in sensitivity. In our model, we find an increased fraction of dividing cells within the JAK2V617F LT-HSCs.
There is support in the literature for the concept that CDK6 regulates apoptosis. In cyclin D3-CDK6–high human tumors, CDK6 inhibits the glycolytic enzymes PFK1 and PKM2 to prevent apoptosis of T-cell acute lymphoblastic leukemia cells.70 Similarly, CDK6 facilitates the survival of FLT3-ITD+ acute myeloid leukemia cells.27 Our data reveal a new mechanism by which CDK6 interferes with apoptosis: in JAK2V617F; Cdk6−/− hematopoietic progenitors, the transcription of the cell-death inducer Klf6, and its mediator Atf3 as well as Btg2, a NF-κB–responsive gene, and Pmaip1, a p53-inducible antiproliferative gene, is significantly enhanced.
MPN is characterized by a chronic state of inflammation. Plasma levels of inflammatory cytokines (eg, IL-1, IL-2, IL-6, IL-8, IL-12, TNF-α, and interferon-γ) are increased and linked to an adverse outcome. NF-κB is a master regulator of inflammation and is constitutively active in malignant and nonmalignant hematopoietic cells in mouse models of MPN.7 CDK6 interacts physically and functionally with the NF-κB subunit p65; the complex is found at the promoters of transcriptionally active NF-κB target genes.8,25 We now add another layer of complexity: in the context of JAK2V617F, Cdk6 promotes NF-κB signaling by suppressing transcription of genes such as NFκbiz, Socs3, Nr4a1/2, Egr1, Klf4, Klf6, and Atf3 that encode inhibitors of NF-κB signaling.49,50,71-76 The tight control of the NF-κB pathway is thus relaxed, promoting inflammation. We propose that this represents one of the mechanisms by which CDK6 interferes with MPN progression.
Inflammatory responses involve cytokines that target the hematopoietic hierarchy at multiple levels. Although interferons or TNFs act on HSCs,77-79 IL-6 acts on MPPs and promotes the production of myeloid cells while reducing the output of the lymphoid lineage.80,81 In a transgenic Bcr/Abl mouse model, IL-6–producing myeloid cells create a pathogenic feed-forward loop where normal and transformed MPPs produce IL-6, which stimulates the production of more myeloid cells.80 Similarly, IL-1 and IL-17 exert myelopoietic effects, although they act on different target cells.82,83 Because IL-1β and IL-6 rely on a CDK6/NF-κB axis in the context of the JAK2V617F mutation, we speculate that the lack of this interaction prevents the expansion of maturing Gr1+/Mac1+ myeloid cells as well as monocytes, eosinophils, and neutrophils in JAK2V617F-positive animals that lack Cdk6. The notion is consistent with the rapid clinical benefits upon administration of IL-1Ra, an antibody-based inhibitor of the receptor bound by the proinflammatory IL-1β.84 Our findings provide a rationale for the therapeutic evaluation of CDK6 shutdown in MPN.
When CDK6 was initially found to regulate transcription, it was initially believed that its transcriptional function was independent of its kinase activity.24,26 Subsequent work revealed that CDK6 also controls the transcription of some genes in a manner that requires its kinase activity.11,27 In the context of the JAK2V617F mutation, CDK6 kinase activity does not contribute to its functions in NF-κB activation, induction of cell death, and HSC kinetics: the proapoptotic effects of loss of the CDK6 protein is not mimicked by palbociclib treatment in primary stem cells from MPN patients.
In summary, our data reveal a critical role for CDK6 in coordinating the activation of HSCs, in promoting prosurvival signals, and in enhancing inflammatory responses in JAK2V617F-mediated MPN, at least partially by direct transcriptional regulation of key genes involved in these processes. CDK6 thereby functions in a manner that is independent of its kinase activity. Our work indicates that fine-tuning of the level of CDK6 could potentially improve the quality of life of MPN patients and thus provides a rationale for the therapeutic use of CDK6-specific degraders based on proteolysis-targeting chimeras.
The data reported in this article have been deposited in the Gene Expression Omnibus database (accession number GSE123401).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Sabine Fajmann, Petra Kudweis, Michael Gurbisz, Bettina Gisslinger, Susanne Herndlhofer, Georg Greiner, and Otto Zach for excellent technical assistance, Peter Bettelheim for sharing patient samples, and Agnieszka Piszczek, Tamara Engelmaier, and Julia Klughofer of the VBCF Histopathology facility for expert histotechnology services and the animal care takers for support. They also thank Graham Tebb for critically revising the manuscript.
This work has received funding from the European Research Council under the European Union’s Horizon 2020 research and innovation program grant agreement 694354 (V.S.) and was also supported by the Austrian Science Fund grants P30041 (H.N.), SFB F4702 (R.K.), F4704 (P.V.), and F4706 (V.S.).
Authorship
Contribution: V.S. was the principal investigator and takes primary responsibility for the article; I.Z.U., B.M., H.N., P.J., K.K., J.D.M.F., M.P.-M., S.L., B.G., and R.H. performed the laboratory work and analyzed data for this study; I.Z.U., B.M., and V.S. wrote the manuscript; P.J., M.Z., and R.G. performed bioinformatics analysis of the RNA sequencing; A.K. analyzed and presented the histopathology data; C.B. performed the RNA sequencing; H.G. and P.V. provided patient samples and the final diagnosis and supervised the laboratory work at the Medical University of Vienna; and R.K. supervised the laboratory work at the CeMM, Vienna and contributed to development of the study.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Veronika Sexl, Institute of Pharmacology and Toxicology, University of Veterinary Medicine, Veterinaerplatz 1, A-1210 Vienna, Austria; e-mail: veronika.sexl@vetmeduni.ac.at.
