

Key Points
Ponatinib therapy can result in VWF-mediated platelet adhesion to the microvascular endothelium.
Ponatinib-related microangiopathy can reduce segmental left ventricular dysfunction.

Abstract
The third-generation tyrosine kinase inhibitor (TKI) ponatinib has been associated with high rates of acute ischemic events. The pathophysiology responsible for these events is unknown. We hypothesized that ponatinib produces an endothelial angiopathy involving excessive endothelial-associated von Willebrand factor (VWF) and secondary platelet adhesion. In wild-type mice and ApoE−/− mice on a Western diet, ultrasound molecular imaging of the thoracic aorta for VWF A1-domain and glycoprotein-Ibα was performed to quantify endothelial-associated VWF and platelet adhesion. After treatment of wild-type mice for 7 days, aortic molecular signal for endothelial-associated VWF and platelet adhesion were five- to sixfold higher in ponatinib vs sham therapy (P < .001), whereas dasatinib had no effect. In ApoE−/− mice, aortic VWF and platelet signals were two- to fourfold higher for ponatinib-treated compared with sham-treated mice (P < .05) and were significantly higher than in treated wild-type mice (P < .05). Platelet and VWF signals in ponatinib-treated mice were significantly reduced by N-acetylcysteine and completely eliminated by recombinant ADAMTS13. Ponatinib produced segmental left ventricular wall motion abnormalities in 33% of wild-type and 45% of ApoE−/− mice and corresponding patchy perfusion defects, yet coronary arteries were normal on angiography. Instead, a global microvascular angiopathy was detected by immunohistochemistry and by intravital microscopy observation of platelet aggregates and nets associated with endothelial cells and leukocytes. Our findings reveal a new form of vascular toxicity for the TKI ponatinib that involves VWF-mediated platelet adhesion and a secondary microvascular angiopathy that produces ischemic wall motion abnormalities. These processes can be mitigated by interventions known to reduce VWF multimer size.
Introduction
Tyrosine kinase inhibitors (TKIs) have revolutionized the treatment of patients with chronic myelogenous leukemia (CML). In CML, the t(9;22) chromosomal translocation results in production of the oncogenic tyrosine kinase BCR-ABL1. TKIs targeted to BCR-ABL1 inhibit the proliferation and survival of malignant cells and result in long-term disease-free survival rates of nearly 80%.1,2 Ponatinib is a third-generation BCR-ABL1 TKI with a broad range of activity against other TKs (vascular endothelial growth factor, fibroblast growth factor, and platelet-derived growth factor receptors) and is the only TKI that is approved for treating CML rendered drug resistant by the T315I mutation.3-5 However, the use of ponatinib in patients has been curtailed, and at one point temporarily halted, by the US Food and Drug Administration, because of high rates of vascular toxicity manifest as acute thrombotic occlusive events within months of initiating therapy, the cause of which is unknown.6,7
In this study, in vivo imaging methods unique in their ability to investigate events at the endothelial-blood pool interface, including in vivo ultrasound molecular imaging and intravital microscopy, were used to assess endothelial alterations that underlie the vascular complications of ponatinib. These approaches have recently been applied to elucidate sudden high-risk endothelial changes that occur globally after focal myocardial ischemia.8 We applied these techniques to investigate whether ponatinib produces an arterial and microvascular endothelial angiopathy involving excessive endothelial-associated von Willebrand factor (VWF) and secondary glycoprotein-Ibα (GPIbα)–mediated platelet adhesion. Because myocardial infarction is one of the most frequent vascular complications, we also assessed whether these endothelial changes result in ischemic left ventricular (LV) wall motion abnormalities (WMAs). With regards to potential mitigation strategies, we tested whether the platelet-related angiopathy could be partially prevented by N-acetylcysteine (NAC), which can potentially reduce VWF multimer size through direct effects on self-association and secondary effects on endogenous protease activity.9
Materials and methods
Animals
The study was approved by the Animal Care and Use Committee of Oregon Health & Science University. We studied wild-type C57Bl/6 mice and hyperlipidemic mice with susceptibility to atherosclerosis produced by gene deletion of the apolipoprotein-E (ApoE−/−) fed a “Western-style” diet (WSD) consisting of 42% of calories from fat (Teklad TD.88137, Envigo) for 6 weeks prior to the study. Mice were studied at 15 to 20 weeks of age, which for ApoE−/− mice on a WSD is expected to represent a stage at which there are early atherosclerotic lesions in the aortic root and proximal aorta.10,11 For all studies except intravital microscopy, mice were anesthetized with 1.0% to 2.0% inhaled isoflurane, and euthermia was maintained. A jugular cannula was placed for IV injection of contrast agents or drugs when necessary. For intravital microscopy, mice were anesthetized with intraperitoneal injection (10-15 μL/g) of a solution containing ketamine hydrochloride (10 mg/mL), xylazine (1 mg/mL), and atropine (0.02 mg/mL).
TKI drug therapy and study design
For TKI therapy in mice, either ponatinib (30 mg/kg) or dasatinib (20 mg/kg) was dissolved in a solution of citrate titrated to achieve a pH of 2.75 and administered as a daily oral gavage. Doses of TKI were selected a priori based on studies demonstrating therapeutic response in murine models of malignancy.12-14 NAC was administered by daily gavage at the same time as ponatinib at a dose (600 mg/kg) that was weight adjusted to be in slight excess to that used to treat acetaminophen poisoning. Mice were treated for 7 days prior to being studied with contrast-enhanced ultrasound (CEU) molecular imaging, intravital microscopy, echocardiography, or computed tomography (CT) coronary angiography. Daily blood pressures were performed (see supplemental Methods, available on the Blood Web site).
Contrast ultrasound molecular imaging study assignments
The following conditions were tested to evaluate aortic endothelial response to TKIs using contrast-enhanced ultrasound as an in vivo readout:
Molecular imaging performed in wild-type mice at baseline and after 7 days of daily therapy with ponatinib, dasatinib, or vehicle.
Molecular imaging performed in ApoE−/− mice on a WSD at baseline and after 7 days of daily therapy with ponatinib, ponatinib + NAC, or vehicle.
Molecular imaging performed in ApoE−/− mice on a WSD after 7 days of daily therapy with ponatinib that also received recombinant human ADAMTS13 (a disintegrin and metalloprotease with thrombospondin type I repeats-13) (5 μg IV) 1 hour prior to imaging. ADAMTS13, which is a regulatory protease that cleaves VWF at the A2 domain, was used to reduce ultralarge VWF multimer size after surface expression by endothelial cells.15,16
Targeted microbubble contrast agent preparation for CEU molecular imaging
Lipid-shelled decafluorobutane microbubbles bearing a bifunctional conjugation moiety consisting of a hydrophobic domain and a biotin domain at the end of a polyethylene glycol spacer were prepared by sonication of a gas-saturated aqueous suspension of distearoylphosphatidylcholine (2 mg/mL), polyoxyethylene-40-stearate (1 mg/mL), and distearoylphosphatidylethanolamine-PEG(2000)biotin (0.4 mg/mL). Conjugation of biotinylated ligand to the microbubble surface was performed with biotin-streptavidin bridging as previously described.11,17 Ligands used for targeting were dimeric murine recombinant A1 domain of VWF A1 (mature VWF amino acids 445 to 716) for targeting platelet GPIbα, and a cell-derived peptide representing the N-terminal 300 amino acids of GPIbα for targeting the VWF A1 domain, which is exposed by shear on ultralarge multimers.18 Control microbubbles were prepared with isotype control antibody (R3-34, BD Biosciences). Microbubble concentrations and size distributions were measured by electrozone sensing (Multisizer III; Beckman Coulter, Brea, CA).
Contrast ultrasound molecular imaging
CEU molecular imaging of the ascending thoracic aorta and proximal arch were imaged using a right parasternal window with a 15L7 linear-array probe (Sequoia; Siemens, Mountain View, CA). Multipulse phase-inversion and amplitude-modulation imaging was performed to specifically detect the nonlinear signals generated at the fundamental and harmonic frequencies by microbubble cavitation.19,20 Imaging was performed at a transmission frequency of 7 MHz and a dynamic range of 55 dB. The mechanical index was set at 1.0. Images were acquired 8 minutes after IV injection of targeted or control microbubbles (1 × 106) performed in random order. Signal from retained microbubbles alone was determined as previously described by acquiring the first ultrasound frame and then digitally subtracting several averaged frames obtained after complete destruction of microbubbles at a mechanical index of 1.4 to eliminate signal from the low concentration of freely circulating microbubbles in the blood pool.11 Signal intensity was measured from a region of interest encompassing the entire ascending aorta to just beyond the origin of the brachiocephalic artery. Region selection was facilitated by fundamental 2-dimensional imaging at 14 MHz acquired after each CEU imaging sequence.
Echocardiography
Echocardiography was performed to assess LV function (see supplemental Methods).
CT coronary angiography
CT coronary angiography was performed immediately after echocardiography in several sham-treated mice and ApoE−/− mice treated with ponatinib that were selected based on presence of obvious WMAs. Mice were treated with 1000 U sodium heparin by intraperitoneal injection. After 15 minutes, an anterior laparotomy was performed in order to selectively cannulate the descending aorta. Retrograde perfusion of the coronary circulation at a perfusion pressure of 100 mm Hg was performed with phosphate-buffered saline at 37°C containing 0.1 mM sodium nitroprusside and 1 mM papaverine in order to clear the intravascular blood pool and achieve maximal vasodilation of the coronary vasculature. Retrograde perfusion fixation was performed with 1% paraformaldehyde followed immediately by perfusion with a radiopaque silicone-based casting agent (Microfil Flow Tech, Carver, MA) prepared as 8:1:1 parts latex/dilutent/curing agent, per the manufacturer’s instructions. Once the coronary arteries were visually confirmed to be filled, perfusion pressure was maintained for an additional 5 to 10 minutes. After curing for 1 hour, the heart was removed and immersion fixed in 2% paraformaldehyde overnight. Micro-CT angiography (Inveon, Siemens) was performed using a 14-bit X-ray detector and a full 360° rotation at 1° increments and 6870-ms exposure times. Imaging was performed at a peak voltage of 30 kV (0.5 mA tube current) with maximal intensity projection processing.
Spatial assessment of perfusion by microspheres
See supplemental Methods.
Intravital microscopy
Direct observation of platelet- and leukocyte-endothelial interactions was performed with intravital microscopy in male wild-type mice and ApoE−/− mice on a WSD after 1 week of treatment with ponatinib or vehicle. The cremaster muscle was exteriorized and prepared for intravital microscopy using a buffered isothermic superfusion as previously described.21 Microscopy was performed with combined fluorescent epi-illumination and low-intensity transillumination (Axioskop2-FS; Carl Zeiss, Thornwood, NY) and a high-resolution charge-coupled device camera (C2400; Hamamatsu Photonics, Hamamatsu, Japan). In vivo fluorescent labeling of platelets was performed with rhodamine-6G (1 mg/mL, 75-100 μL IV). The number of adherent leukocytes and platelet adhesive events (>3 s) and the formation of platelet “strings” indicative of ultra-large VWF multimers were quantified as either the number of events or the fluorescent platelet area normalized to vessel area. A minimum of 5 separate venules were assessed per animal.
Flow cytometry
Flow cytometry was used to assess platelet surface adhesion molecule expression (see supplemental Methods).
Endothelial ROS production, viability, and VWF secretion
Murine endothelial cells (SVEC4-10; ATCC, Manassas, VA) were grown to confluence in Dulbecco’s modified Eagle medium growth medium supplemented with 10% fetal bovine serum. Cells were exposed to ponatinib added to the growth medium to reach a final concentration of 0.1, 0.5, 1.0, or 2.5 μM for 2 hours. Positive control experiments were performed by adding 10 mL of a 100 mM solution of phorbol 12-myristate 13-acetate to SVE4-10 cells for 30 minutes. For assessing reactive oxygen species (ROS), 2′,7′-dichlorodihydrofluorescein diacetate succinimidyl ester (H2DCFDA; OxyBURST Green, ThermoFisher Scientific) was added to the culture medium at a final concentration of 1 mM for 10 minutes prior to fluorescence microscopy (Axioscop-2, Carl Zeiss) at an emission wavelength of 490 nm and a constant output intensity. Mean fluorescent intensity for H2DCFDA was measured from a minimum of 10 random fields of view (ImageJ; National Institutes of Health, Bethesda, MD).22 For cell viability, costaining was performed with propidium iodide (PI) at a final concentration of 5 mM and 4′,6-diamidin-2-phenylindol (DAPI) for 5 minutes then washed. Endothelial cell viability was measured by the proportion of DAPI-stained nuclei that also stained with PI in a minimum of 10 randomly selected fields of view. For assessment of VWF secretion, a microfluidic cell culture chamber (VI0.1; ibidi, Martinsried, Germany) was seeded with 2 × 106 human umbilical vein endothelial cells (HUVECs) and infused with complete medium overnight at a shear rate of 200 s−1. Cells were exposed to serum-free medium for 2 hours and then for 24 hours with serum-free medium with either 0.1% fatty acid-free bovine serum albumin (control), ponatinib (0.5 mM), or tumor necrosis factor α (TNF-α; 10 ng/mL) (positive control). Cells were then fixed and immunostained without permeabilization with rabbit anti-human VWF (A0082; DAKO, Santa Clara, CA) and mouse anti-human CD31 (WM59; Thermo Fisher Scientific, Grand Island, NY) polyclonal antibodies, and Alexa Fluor–labeled secondary antibodies. Area staining for VWF was assessed in 5 random fields of view per chamber.
Histology
For immunohistochemistry of the LV myocardium, myocardial blood volume was removed by retrograde aortic perfusion at physiologic pressure of isothermic phosphate-buffered saline containing 2.5% albumin. Perfusion-fixed short-axis thick sections that, for ponatinib-treated mice, included both the areas with and without WMA were cut. Immersion-fixed sections from the kidneys of ponatinib- and vehicle-treated mice were also assessed. Immunohistochemistry was performed using rat anti-mouse monoclonal antibody against platelet CD41 (ab33661; Abcam, Cambridge, MA), and secondary staining with donkey ant-rat Cy3-labeled polyclonal antibody (Jackson ImmunoResearch, West Grove, PA). Endothelial staining was performed with Alexa Fluor 488–conjugated isolectin GS-IB4 (Invitrogen, Grand Island, NY). Nuclear counterstaining was performed with Hoechst 33342 (Invitrogen). Fluorescent microscopy was performed on a confocal system (TCS SP5; Leica Microsystems, Buffalo Grove, IL).
Statistical analysis
See supplemental Methods for statistical analysis.
Results
Survival and blood pressure in ponatinib-treated mice
Because clinical studies have indicated that patients with traditional atherosclerotic risk factors are at greater risk for ponatinib vascular toxicity,7 we studied both wild-type C57Bl/6 mice and mice with gene-targeted deletion of ApoE−/− fed for 6 weeks with WSD. During the 1-week period of therapy, treatment-related mortality occurred only in ponatinib-treated mice, with a significantly higher mortality compared with sham-treated animals for the ApoE−/− mice (Figure 1). Daily awake tail cuff blood pressure measurements in animals acclimatized to the procedure revealed a gradual increase in both systolic and diastolic blood pressure in ponatinib-treated wild-type and ApoE−/− mice, whereas sham-treated mice remained normotensive (Figure 1; supplemental Figure 1). This hypertensive response is similar to blood pressure responses seen in two-thirds of patients receiving ponatinib23 and likely reflects potency of ponatinib kinase against vascular endothelial growth factor receptor-2. Blood counts after days of therapy showed mild leukocytosis in ponatinib-treated mice (supplemental Table 1). Schistocytes were not detected using Wright-Giemsa stain (supplemental Figure 1).

Survival and blood pressure according to treatment assignments. Kaplan-Meier curves illustrate survival after initiation of ponatinib (30 mg/kg per day) or vehicle (sham treatment) in wild-type C57Bl/6 mice (A) and ApoE−/− mice on a WSD (B). Tail-cuff systolic blood pressure was measured in awake wild-type (C) and ApoE−/− mice on a WSD (D) in animals that were acclimatized to the procedure prior to initiation of therapy. *P < .05 vs vehicle. BP, blood pressure.
Survival and blood pressure according to treatment assignments. Kaplan-Meier curves illustrate survival after initiation of ponatinib (30 mg/kg per day) or vehicle (sham treatment) in wild-type C57Bl/6 mice (A) and ApoE−/− mice on a WSD (B). Tail-cuff systolic blood pressure was measured in awake wild-type (C) and ApoE−/− mice on a WSD (D) in animals that were acclimatized to the procedure prior to initiation of therapy. *P < .05 vs vehicle. BP, blood pressure.
Vascular prothrombotic changes by molecular imaging
Molecular imaging was performed with tracers targeted to endothelial-associated large multimers of VWF that have undergone shear-mediated exposure of their platelet binding domain and to platelet GPIbα, which reflects vascular platelet adhesion. In wild-type mice, aortic signal for VWF-A1 and platelet GPIbα in ponatinib-treated mice was five- to sixfold greater compared with sham-treated mice (Figure 2A-B). This high degree of signal for VWF and platelet adhesion was not seen after therapy with dasatinib (20 mg/kg per day), a second-generation TKI characterized by a narrower spectrum activity than ponatinib with regards to non–BCR-ABL1 receptors that has been shown to have a much lower rate of vascular events in patients.5,6 Flow cytometry performed with platelets from vehicle-treated vs ponatinib-treated mice showed no difference in surface expression for GPIbα (median fluorescence intensity, 9156 ± 1439 vs 9505 ± 1684, P = .90) or P-selectin (median fluorescence intensity, 6992 ± 860 vs 7536 ± 1291, P = .70).
![Figure 2. Molecular imaging of aortic endothelial phenotype in wild-type and ApoE−− mice. (A) Mean (± standard error of the mean [SEM]) signal enhancement measured from the proximal thoracic aorta on CEU molecular imaging using tracers targeted to platelet GPIbα, VWF A1-binding domain, or control agent, in wild-type mice treated for 1 week with ponatinib (30 mg/kg per day), dasatinib (20 mg/kg per day), or vehicle. *P < .01. (B) Illustrative images from a ponatinib-treated wild-type mouse showing 2-dimensional (2-D) ultrasound (14 MHz) of the proximal thoracic aorta and origin of the brachiocephalic artery (outlined), and background-subtracted color-coded (scales at right) CEU molecular imaging with control or targeted contrast agents. (C) Mean (± SEM) signal enhancement on CEU molecular imaging of the proximal thoracic aorta in ApoE−/− mice on a WSD treated for 1 week with ponatinib (30 mg/kg per day) or vehicle. *P < .05. (D) CEU molecular imaging in ponatinib-treated ApoE−/− mice on a WSD showing effects of either daily coadministration of NAC (600 mg/kg per day) or IV rADAMTS13 (5 μg) given 1 hour prior to imaging. *P < .05; **P < .01. IU, international unit.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/133/14/10.1182_blood-2018-10-881557/4/m_blood881557f2.png?Expires=1769109510&Signature=FBPwiIHryuuTkttagHIHXuI5tdiBvwKT46U8OxyqnxttXaxEKGIt5s~gVLZPtaTO3yvN2-azMFT-yeDO1NCqu-dtWCG5o1timoCz~A6-NyNNW0eNihZxuOExzl4YRVSJoPfYRu3mrbf16NRzOeUx128NMexoWdeTf4X5EYDz2ZolznKkC6tX4ClR~RPHtoAvAMd0Y-NQzHSuwHVZotug-8BSmsqGisxSD8XVz4phloyIyztXnbnshhNB73aJEo8VtsjpL1kHg-qSe8iMiPQe~ME88UdmCepCZEHb~ZjVJ~HNMgCgQ60VtO2z235VuimZ0MHmIQzinrbLpwuc6yprDA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Molecular imaging of aortic endothelial phenotype in wild-type and ApoE−− mice. (A) Mean (± standard error of the mean [SEM]) signal enhancement measured from the proximal thoracic aorta on CEU molecular imaging using tracers targeted to platelet GPIbα, VWF A1-binding domain, or control agent, in wild-type mice treated for 1 week with ponatinib (30 mg/kg per day), dasatinib (20 mg/kg per day), or vehicle. *P < .01. (B) Illustrative images from a ponatinib-treated wild-type mouse showing 2-dimensional (2-D) ultrasound (14 MHz) of the proximal thoracic aorta and origin of the brachiocephalic artery (outlined), and background-subtracted color-coded (scales at right) CEU molecular imaging with control or targeted contrast agents. (C) Mean (± SEM) signal enhancement on CEU molecular imaging of the proximal thoracic aorta in ApoE−/− mice on a WSD treated for 1 week with ponatinib (30 mg/kg per day) or vehicle. *P < .05. (D) CEU molecular imaging in ponatinib-treated ApoE−/− mice on a WSD showing effects of either daily coadministration of NAC (600 mg/kg per day) or IV rADAMTS13 (5 μg) given 1 hour prior to imaging. *P < .05; **P < .01. IU, international unit.
Molecular imaging of aortic endothelial phenotype in wild-type and ApoE−− mice. (A) Mean (± standard error of the mean [SEM]) signal enhancement measured from the proximal thoracic aorta on CEU molecular imaging using tracers targeted to platelet GPIbα, VWF A1-binding domain, or control agent, in wild-type mice treated for 1 week with ponatinib (30 mg/kg per day), dasatinib (20 mg/kg per day), or vehicle. *P < .01. (B) Illustrative images from a ponatinib-treated wild-type mouse showing 2-dimensional (2-D) ultrasound (14 MHz) of the proximal thoracic aorta and origin of the brachiocephalic artery (outlined), and background-subtracted color-coded (scales at right) CEU molecular imaging with control or targeted contrast agents. (C) Mean (± SEM) signal enhancement on CEU molecular imaging of the proximal thoracic aorta in ApoE−/− mice on a WSD treated for 1 week with ponatinib (30 mg/kg per day) or vehicle. *P < .05. (D) CEU molecular imaging in ponatinib-treated ApoE−/− mice on a WSD showing effects of either daily coadministration of NAC (600 mg/kg per day) or IV rADAMTS13 (5 μg) given 1 hour prior to imaging. *P < .05; **P < .01. IU, international unit.
In ApoE−/− mice on a WSD, ponatinib treatment was again associated with a much higher aortic molecular imaging signal for VWF-A1 and platelet GPIbα compared with sham-treated animals (Figure 2B). Compared with wild-type mice, ApoE−/− mice had a higher aortic molecular imaging signal after ponatinib for both platelet GPIbα (median [interquartile range]: 25.1 [21.0-32.3] vs 13.3 [10.6-21.2], P = .02) and for VWF-A1 (25.1 [18.1-31.2] vs 15.3 [11.8-18.9], P = .02). In ponatinib-treated ApoE−/− mice, IV administration of recombinant ADAMTS13 1 hour prior to molecular imaging resulted in nearly complete elimination of aortic VWF and GPIbα signal enhancement (Figure 2D), indicating that platelet adhesion was entirely attributable to endothelium-associated VWF. Treatment of ApoE−/− mice with NAC, coadministered daily with ponatinib, also eliminated the VWF signal but reduced the platelet signal only by half.
Myocardial function and coronary patency
Because cardiac events are among the most common of the reported ponatinib vascular toxicities seen in humans,7 transthoracic 2-dimensional echocardiography was performed to assess for ischemic WMAs. LV function was normal in all animals at baseline and remained normal after sham treatment in wild-type and ApoE−/− mice. In ponatinib-treated mice, new segmental WMAs were frequently observed (Figure 3A-B; supplemental Videos 1-3). Segmental WMAs resulted in a decrease in average stroke volumes and, for ApoE−/− mice, a worsening in systolic global radial strain (Figure 3C-D; supplemental Figure 2). Coadministration of NAC in ponatinib-treated ApoE−/− mice prevented the reduction in stroke volume and global radial strain and reduced the proportion of animals with abnormal wall motion at 1 week, although the latter did not meet statistical significance (Fisher’s exact P = .20). In mice with WMAs, spatially matched patchy LV myocardial perfusion defects were observed on postmortem analysis of the distribution of fluorescent microspheres given just prior to euthanasia (Figure 4A). Quantitatively, the area void of microspheres was greater for ponatinib-treated than sham-treated animals for both wild-type (23.1 ± 11.7 vs 3.1 ± 2.2, P < .001) and ApoE−/− mice (26.5 ± 9.3 vs 5.7 ± 2.7, P < .001). Despite the presence of perfusion defects on microsphere analysis, CT coronary arteriography failed to detect coronary artery stenosis or occlusion in ponatinib-treated mice (Figure 4B). To investigate the possibility of a microvascular thrombotic angiopathy, immunohistochemistry of the LV was performed and showed multiple large platelet aggregates in ponatinib-treated mice only in myocardial regions with WMAs (Figure 4C-D). Aggregates were absent in sham-treated mice. Immunohistochemistry of renal tissue, which was performed to assess a nonmyocardial organ, also showed an increase in microvascular platelets in the form of small aggregates (supplemental Figure 3).

Echocardiographic detection of LV dysfunction. (A) Echocardiography in the parasternal long-axis plane at end-diastole and end-systole illustrating an inferoapical WMA (arrow, see online videos for examples of WMAs). (B) Proportion of animals with segmental LV wall motion abnormalities after 1 week of ponatinib therapy. (C-D) Echocardiographic measurement of stroke volume (bars represent mean ± standard deviation) at baseline and after treatment (vehicle or ponatinib) in wild-type (C) and ApoE−/− mice on a WSD (D). ns, not significant.
Echocardiographic detection of LV dysfunction. (A) Echocardiography in the parasternal long-axis plane at end-diastole and end-systole illustrating an inferoapical WMA (arrow, see online videos for examples of WMAs). (B) Proportion of animals with segmental LV wall motion abnormalities after 1 week of ponatinib therapy. (C-D) Echocardiographic measurement of stroke volume (bars represent mean ± standard deviation) at baseline and after treatment (vehicle or ponatinib) in wild-type (C) and ApoE−/− mice on a WSD (D). ns, not significant.
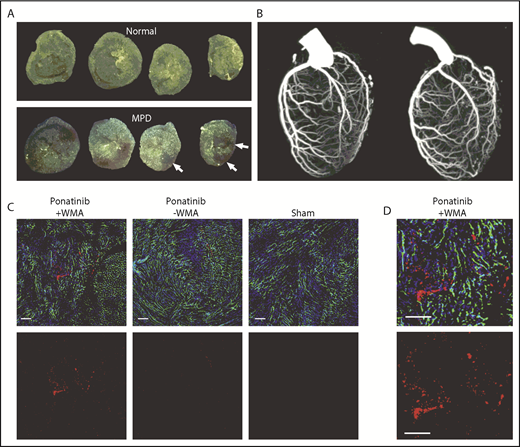
Coronary artery and LV coronary microvascular anatomy. (A) Ex vivo epifluorescent illumination of microspheres in sequential ventricular short-axis sections illustrating normal perfusion, and focal regions of myocardial perfusion defect (MPD) (arrows). (B) CT coronary angiography in the left lateral projection illustrating lack of arterial occlusion in 2 ponatinib-treated mice with WMAs. (C) Fluorescent confocal microscopy of the LV myocardium from ponatinib-treated wild-type mice showing regions with and without WMAs and from a sham-treated mouse. Staining was performed with isolectin (green) for microvessels, Hoechst stain for nuclei (blue), and platelet CD41 immunohistochemistry (red). The bottom rows illustrate the red channel alone to better display platelets. Scale bars, 100 μm. (D) Higher-magnification image from the ponatinib-treated animal with WMA.
Coronary artery and LV coronary microvascular anatomy. (A) Ex vivo epifluorescent illumination of microspheres in sequential ventricular short-axis sections illustrating normal perfusion, and focal regions of myocardial perfusion defect (MPD) (arrows). (B) CT coronary angiography in the left lateral projection illustrating lack of arterial occlusion in 2 ponatinib-treated mice with WMAs. (C) Fluorescent confocal microscopy of the LV myocardium from ponatinib-treated wild-type mice showing regions with and without WMAs and from a sham-treated mouse. Staining was performed with isolectin (green) for microvessels, Hoechst stain for nuclei (blue), and platelet CD41 immunohistochemistry (red). The bottom rows illustrate the red channel alone to better display platelets. Scale bars, 100 μm. (D) Higher-magnification image from the ponatinib-treated animal with WMA.
Microvascular platelet adhesion
To further assess for global thrombotic microangiography, intravital microscopy of the cremaster muscle was used to directly observe the microcirculation for similar endothelial-related changes. In wild-type mice and in ApoE−/− mice on a WSD, ponatinib resulted in a greater number of platelet–endothelial interactions manifest either as nontransient platelet endothelial adhesion or as platelet aggregates in the form of linear strings and nets associated with the endothelium or leukocytes (Figure 5; supplemental Videos 4-8). Events were seen in both venules and arterioles, and occasional embolization was observed (supplemental Video 7). Because platelet adhesion can promote endothelial cell adhesion molecular expression and can directly participate in leukocyte adhesion and extravasation,24,25 leukocyte recruitment was also analyzed. Ponatinib-treated mice had substantially slower venular leukocyte rolling velocities, but only trends for greater leukocyte adhesion were seen (Figure 5). Plasma showed modest, nonsignificant increases in VWF antigen levels by enzyme-linked immunosorbent assay for ponatinib-treated wild-type and ApoE−/− mice compared with sham-treated controls (supplemental Figure 4).
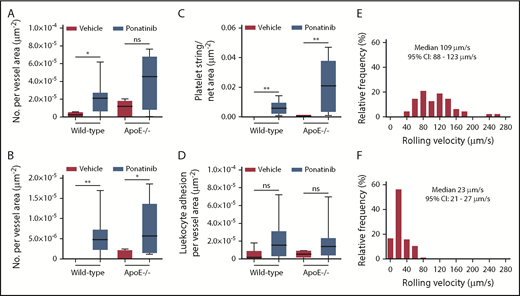
Microvascular platelet and leukocyte recruitment on intravital microscopy. Box-whisker plots for intravital microscopy data from the cremasteric microcirculation of vehicle (sham) or ponatinib-treated wild-type and ApoE−/− mice illustrating the number of platelets adhering to the microvascular endothelium (A), the number of platelet strings or nets (B), the area of adherent platelet strings or nets (C), and the number of leukocytes adhered in postcapillary venules (D). *P < .05; **P < .01. (E-F) Histograms illustrating the distribution of leukocyte rolling velocities in cremasteric venules for ApoE−/− mice on a WSD for animals treated with vehicle (E) or ponatinib (F). Rolling velocity was slower for ponatinib-treated mice (P < .01 by Mann-Whitney rank-sum test). ns, not significant.
Microvascular platelet and leukocyte recruitment on intravital microscopy. Box-whisker plots for intravital microscopy data from the cremasteric microcirculation of vehicle (sham) or ponatinib-treated wild-type and ApoE−/− mice illustrating the number of platelets adhering to the microvascular endothelium (A), the number of platelet strings or nets (B), the area of adherent platelet strings or nets (C), and the number of leukocytes adhered in postcapillary venules (D). *P < .05; **P < .01. (E-F) Histograms illustrating the distribution of leukocyte rolling velocities in cremasteric venules for ApoE−/− mice on a WSD for animals treated with vehicle (E) or ponatinib (F). Rolling velocity was slower for ponatinib-treated mice (P < .01 by Mann-Whitney rank-sum test). ns, not significant.
Endothelial cell ROS production and VWF expression
To better understand the ameliorative effects of NAC in ponatinib-treated mice, production of ROS from ponatinib-treated cultured SVEC4-10 murine endothelial cells was measured. Ponatinib added to the growth media for 24 hours resulted in a concentration-dependent increase in endothelial-derived ROS, as measured by H2DCFDA fluorescence, and a loss of cell confluence at supraphysiologic (>0.5 μM) concentrations (Figure 6).26 There was also a dose-dependent loss of cell viability, as determined by PI staining. The proportion of nonviable cells was substantially attenuated by addition of NAC. Microfluidic chamber experiments in HUVECs exposed to ponatinib (0.5 μM) for 24 hours demonstrated that VWF mobilization to the cell surface was much greater than in control conditions and was equivalent to that achieved by TNF-α (Figure 6).
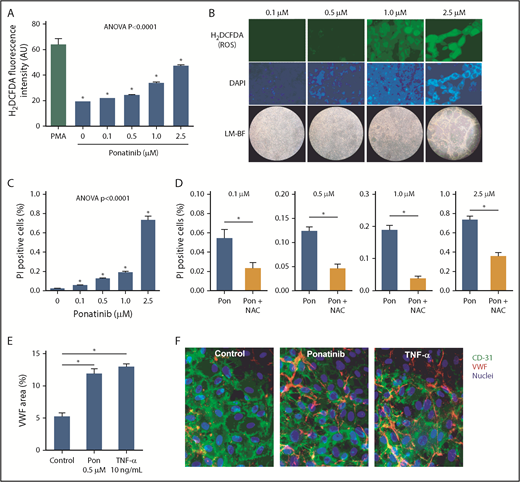
Dose-dependent effects of ponatinib on endothelial cell ROS production and viability. (A) ROS generation by SVEC4-10 endothelial cells in culture measured by mean (± SEM) H2DCFDA fluorescence after 24 hours of ponatinib treatment. Positive control data are shown for phorbol 12-myristate 13-acetate (PMA)–treated cells. (B) Examples of SVEC4-10 cells exposed for 24 hours to ponatinib (0.1 to 2.5 μM) by fluorescent imaging of H2DCFDA and DAPI (nuclear staining) illustrating dose-dependent increase in ROS, and low-magnification bright-field (LM-BF) microscopy illustrating loss of cell confluence at high concentrations. (C) Mean (± SEM) cell viability after 24 hours of exposure to ponatinib (0.1 to 2.5 μM) determined by the proportion of cells staining positive for PI. *P < .05 vs all other conditions by post hoc testing after correction for multiple comparisons. (D) Cell viability at various concentrations of ponatinib (Pon) with and without NAC. *P < .05. (E-F) Mean (± SEM) area for externalized VWF on HUVECs in a microfluidic chamber after 24 hours of exposure to ponatinib (0.5 μM), TNF-α (10 ng/mL), or control serum, and examples of fluorescent microscopy for each condition. *P < .01. AU, arbitrary unit.
Dose-dependent effects of ponatinib on endothelial cell ROS production and viability. (A) ROS generation by SVEC4-10 endothelial cells in culture measured by mean (± SEM) H2DCFDA fluorescence after 24 hours of ponatinib treatment. Positive control data are shown for phorbol 12-myristate 13-acetate (PMA)–treated cells. (B) Examples of SVEC4-10 cells exposed for 24 hours to ponatinib (0.1 to 2.5 μM) by fluorescent imaging of H2DCFDA and DAPI (nuclear staining) illustrating dose-dependent increase in ROS, and low-magnification bright-field (LM-BF) microscopy illustrating loss of cell confluence at high concentrations. (C) Mean (± SEM) cell viability after 24 hours of exposure to ponatinib (0.1 to 2.5 μM) determined by the proportion of cells staining positive for PI. *P < .05 vs all other conditions by post hoc testing after correction for multiple comparisons. (D) Cell viability at various concentrations of ponatinib (Pon) with and without NAC. *P < .05. (E-F) Mean (± SEM) area for externalized VWF on HUVECs in a microfluidic chamber after 24 hours of exposure to ponatinib (0.5 μM), TNF-α (10 ng/mL), or control serum, and examples of fluorescent microscopy for each condition. *P < .01. AU, arbitrary unit.
Discussion
This study provides evidence for a new form of cardiotoxicity associated with TKIs that are used for therapy in CML. Our results indicate that the third-generation TKI ponatinib, which has a broad spectrum of TKI activity and is active against disease that is drug resistant from the T315I mutation, can produce prothrombotic angiopathy manifest by platelet adhesion. This platelet adhesion is mediated by GPIbα binding of exposed A1 domain on VWF that is endothelial associated or associated with leukocyte–platelet complexes. These processes occur in both large arteries and the peripheral microcirculation and in the latter can produce ischemic changes in ventricular function. These platelet-mediated events are likely to be involved in the processes responsible for the high incidence of vascular toxicity associated with ponatinib in patients with CML.6,7,27
Previous studies investigating potential mechanisms for ischemic events with ponatinib have been largely inconclusive. A knowledge gap existed regarding the potential role of the endothelium. To investigate this possibility, we applied CEU molecular imaging, which employs tracers that are confined to the vascular compartment to assess endothelial events.8,18 Molecular imaging of the proximal thoracic aorta, a site that is susceptible to atherosclerosis in WSD-fed ApoE−/− mice,11 was performed with tracers targeted to (1) endothelial-associated large multimers of VWF that have undergone shear-mediated exposure of the A1-binding domain for the GPIbα component of the GPIb-IX-V complex on platelets or (2) GPIbα, which reflects platelet adhesion to the endothelium and is constitutively expressed by platelets.8,18 This approach has recently been used to examine similar global endothelial abnormalities that occur after acute myocardial infarction that are responsible for a sudden increase in events in nonculprit arteries.8 Our data indicated that ponatinib increases endothelial VWF with exposure of the A1-binding domain and platelet adhesion several fold within days of initiation. These changes were not seen with the BCR-ABL1–targeted drug dasatinib, which does not have as broad TKI effects as ponatinib and has been associated with much lower rates of thrombotic events.6 While our data and those from prior studies studying ponatinib’s effects on platelets have demonstrated either neutral or inhibitory effects,28,29 the GPIbα-mediated pathways uncovered in this study do not require platelet activation. They would therefore not necessarily respond favorably to traditional antiplatelet therapies used in atherosclerotic disease.
On molecular imaging, IV administration of ADAMTS13, a regulatory zinc protease that cleaves VWF at the Tyr1605-Met1606 site of the A2 domain to regulate multimer size,15,30 eliminated the signal for both VWF and platelets. The reversal of aortic platelet adhesion by high-dose rADAMTS13 therapy in ponatinib-treated animals that had normal ADAMTS13 levels supports the notion that ponatinib causes an acquired resistance to ultralarge multimers of VWF (UL-VWF) proteolytic cleavage that can be overcome with excess enzyme. In addition, ponatinib also increased surface expression of VWF on HUVECs cultured in a microfluidic system, indicating that increased surface mobilization and decreased proteolytic cleavage played a role.
Oxidative modification of ADAMTS13 and VWF can impair cleavage of endothelial UL-VWF31,32 and has been implicated in platelet–endothelial adhesion in atherosclerosis and acquired forms of thrombotic thrombocytopenic purpura.8,33 Accordingly, we tested the effect of coadministration of NAC (600 mg/kg per day by gavage), which maintains antioxidant activity, thereby improving endogenous protease activity.34 We found a significant reduction in both endothelial-associated VWF and platelet adhesion. Because NAC can also directly reduce endothelial-associated UL-VWF through disulfide bond reduction,9 certainty of the role of ROS would require the administration of compounds that influence only oxidative stress.
In ponatinib-treated mice, we observed perfusion defects and regional WMAs without coronary artery occlusion or stenosis. This constellation of findings is consistent with a thrombotic microangiography, which is known to occur in the kidney with vascular endothelial growth factor receptor TKIs.35,36 Thrombotic microangiopathy in diseases such as thrombocytopenic purpura has been associated with high rates of myocardial infarction.37,38 A high proportion of these patients do not have obstructive epicardial coronary artery disease but do have postmortem findings of coronary microvascular thrombosis.37,39 The notion that segmental LV dysfunction and matching perfusion defects without coronary artery abnormalities in ponatinib-treated mice were due to microvascular angiopathy was supported by histologic evidence of platelet adhesio and by direct observation of endothelial-associated platelet strings and nets on intravital microscopy. The latter technique also revealed that platelet networks were attached on the downstream side of leukocytes. Potential reasons for this finding include that platelets bind to neutrophil-associated VWF or neutrophil extracellular traps,40 although parsing these processes would be difficult based on the ability of platelets to trigger neutrophil extracellular traps through TLR-4 signaling.41 The observation of venular embolization of platelet aggregates on intravital microscopy is intriguing, since venous thromboembolism, which involves VWF-mediated platelet adhesion,42 has also been observed in clinical experience. The absence of classic markers of thrombotic microangiopathy such as schistocytes can best be explained by the severity of angiopathy, which was not fulminant, severe, and diffuse but rather indolent and patchy.
There are several limitations of the study that should be mentioned. The short duration of the study was adequate for assessing pathophysiologic mechanisms, but not necessarily all of the clinical sequelae. In humans, hypertension develops rapidly, similar to what we found in mice, but thrombotic complications occur over the course of months. The short duration of our experiments was also not designed to evaluate the worsening of atherosclerosis and thrombotic complications in large arteries seen in clinical experience.6,7 The short-duration protocols we used were not, however, designed to test this possibility. In fact, we believe large- and small-vessel pathologies are likely to be intertwined, since VWF-mediated platelet–endothelial interactions in atherosclerosis promote acceleration of plaque growth and inflammation.8,43 Because of limited blood sampling, we were not able to accurately measure endogenous ADAMTS13 activity, which would have strengthened the weight of our evidence. These measurements may be best suited to prospective study in clinical trials, since human ADAMTS13 assays are well established. Although we demonstrated that ponatinib influences endothelial cell ROS production and viability, we cannot at this time directly link these findings to the dose dependency of arterial thrombotic events. Finally, we studied a murine model lacking BCR-ABL1 without comparison with BCR-ABL1 transgenic models, implying that the thrombotic effects of ponatinib are not mediated through BCR-ABL1 inhibition.
In aggregate, our findings help elucidate mechanisms for vascular toxicity with the BCR-ABL1 TKI ponatinib that can be tested in humans. The identification of VWF-mediated platelet adhesion and a secondary indolent microvascular angiopathy in mice treated with ponatinib provides a basis for future testing in humans and reveals opportunities for potential mitigating strategies.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This work was supported by National Institutes of Health, National Heart, Lung, and Blood Institute grants R01-HL078610 and R01-HL120046, and National Institutes of Health, Office of the Director grant P51-OD011092 (J.R.L.), National Institutes of Health, National Heart, Lung, and Blood Institute grant K08-HL133493 (M.W.), National Institutes of Health, National Heart, Lung, and Blood Institute grants HL42846 and HL78784 (Z.R.), and National Institutes of Health, National Heart, Lung, and Blood Institute grants R01-HL091153 and R01-HL11763 (J.A.L.), the Swiss National Science Foundation (grant P2BSP3-158853) (F.M.), a Japan Society for the Promotion of Science Overseas Research Fellowship (K.O.), and the Manpei Suzuki Diabetes Foundation and Novartis (F.M.). The study was supported by a material grant from Takeda Pharmaceutical Co., which provided ponatinib.
Authorship
Contribution: J.R.L., J.M., O.J.M., and J.A.L. conceived the hypothesis and designed the study; Y.L., F.M., M.W., A.X., W.P., Y.Q., K.O., W.S., E.B., Z.R., J.C., T.S., J.A.L., and J.R.L. performed research and interpreted data; Y.L., F.M., and J.R.L. made the figures; and Y.L., J.M., B.J.D., J.A.L., and J.R.L. wrote the manuscript.
Conflict-of-interest disclosure: B.J.D. is a member of the scientific advisory board (SAB) for Aileron Therapeutics, ALLCRON, Cepheid, Gilead Sciences, Vivid Biosciences, Celgene, and Baxalta (inactive); is an SAB member and owns stock in Aptose Biosciences, Blueprint Medicines, β Cat, GRAIL, Third Coast Therapeutics, and CTI BioPharma (inactive); is a scientific founder of and owns stock in MolecularMD; is a member of the board of directors and owns stock in Amgen; is a member of the board of directors for Burroughs Wellcome Fund, CureOne; is a member of the Joint Steering Committee at Beat AML LLS; and receives clinical trial funding from Novartis, Bristol-Myers Squibb, and Pfizer, and royalties from patent 6958335 (Novartis exclusive license) and Oregon Health & Science University and Dana-Farber Cancer Institute (one Merck exclusive license). The remaining authors declare no competing financial interests.
Correspondence: Jonathan R. Lindner, Knight Cardiovascular Institute, UHN-62, Oregon Health & Science University, 3181 SW Sam Jackson Park Rd, Portland, OR 97239; e-mail: lindnerj@ohsu.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal