

Key Points
FLT3 activation confers ATRA resistance in mouse models of APL.
Arsenic trioxide circumvents ATRA resistance in this setting.

Abstract
Acute promyelocytic leukemia (APL) is often associated with activating FLT3 signaling mutations. These are highly related to hyperleukocytosis, a major adverse risk factor with chemotherapy-based regimens. APL is a model for oncogene-targeted therapies: all-trans retinoic acid (ATRA) and arsenic both target and degrade its ProMyelocytic Leukemia/Retinoic Acid Receptor α (PML/RARA) driver. The combined ATRA/arsenic regimen now cures virtually all patients with standard-risk APL. Although FLT3-internal tandem duplication (ITD) was an adverse risk factor for historical ATRA/chemotherapy regimens, the molecular bases for this effect remain unknown. Using mouse APL models, we unexpectedly demonstrate that FLT3-ITD severely blunts ATRA response. Remarkably, although the transcriptional output of initial ATRA response is unaffected, ATRA-induced PML/RARA degradation is blunted, as is PML nuclear body reformation and activation of P53 signaling. Critically, the combination of ATRA and arsenic fully rescues therapeutic response in FLT3-ITD APLs, restoring PML/RARA degradation, PML nuclear body reformation, P53 activation, and APL eradication. Moreover, arsenic targeting of normal PML also contributes to APL response in vivo. These unexpected results explain the less favorable outcome of FLT3-ITD APLs with ATRA-based regimens, and stress the key role of PML nuclear bodies in APL eradication by the ATRA/arsenic combination.
Introduction
Acute promyelocytic leukemia (APL) is caused by the t(15;17) translocation, which involves the ProMyelocytic Leukemia (PML) and Retinoic Acid Receptor α (RARA) genes.1,2 PML/RARA expression drives leukemogenesis by deregulating differentiation and self-renewal of myeloid progenitors.3 In multiple animal transgenic or knock-in models, PML/RARA expression yields typical APL, although at variable penetrance.4-7
Retinoic acid (RA) receptor α is a transcription factor activated by All-Trans Retinoic Acid (ATRA).8 Some RARA targets regulate myeloid differentiation and granulopoiesis.9 PML is a nuclear protein that forms speckles known as PML nuclear bodies (NBs). These dynamic structures recruit p53 and most of its regulators.10-12 PML NBs are believed to facilitate posttranslational modifications, notably sumoylation and ubiquitinylation,13-15 and play a key role in senescence through their control of p53 and E2F signaling.14,16 The PML/RARA fusion protein binds corepressors and histone deacetylases,17-21 opposing RA-accelerated myeloid maturation. PML/RARA may sometimes activate transcription of some of its primary targets.22 In addition, PML/RARA heterodimerizes with PML, causing the disruption of PML NBs and redistribution of PML in a microspeckled pattern.23-25 This presumably abrogates PML-controlled pathways, and may thus also contribute to APL pathogenesis.26-28
Patients with APL treated with chemotherapy alone had an overall poor prognosis. The addition of ATRA to anthracycline-based chemotherapy was a decisive advance in APL therapy, dramatically increasing complete remission rates and cures.29 Initially, the therapeutic effect of ATRA was believed to reflect its ability to reverse transcriptional repression of myeloid differentiation genes. Yet experiments carried out in APL mice have suggested that blast differentiation may be uncoupled from loss of leukemia-initiating cells self-renewal,30-32 shedding a renewed interest on PML/RARA degradation on ATRA treatment.32-35 Indeed, ATRA-initiated PML/RARA proteolysis allows PML NB reassembly, triggering p53 activation and senescence-like features, and ultimately drives elimination of leukemia-initiating cells.36,37
Another therapy, arsenic trioxide, targets the PML moiety of PML/RARA, inducing its degradation and driving NB association.33,38,39 Arsenic is an extremely efficient and highly selective APL drug that cures up to 70% of patients with APL as a single agent.40,41 The molecular and biochemical bases for arsenic activity involve direct binding onto a specific site in PML driving NB formation and conjugations by Small Ubiquitin-like MOdifier (SUMO), followed by ubiquitin and proteasome-mediated PML/RARA degradation (reviewed in de Thé et al42 and Dos Santos et al43 ). Recently, the frontline combination of ATRA and arsenic in non–high-risk APL allowed the cure of most patients without the need for DNA-damaging chemotherapy.44-48
Internal tandem duplication (ITD) of the fms-like tyrosine kinase 3 gene (FLT3) occurs with a much higher frequency in APL (35%-40%) than in other AMLs (20%-25%).49-56 FLT3 activation is tightly associated with a high white blood cell count at presentation, cases designated as high-risk APL. Although hyperleukocytosis is a well-known adverse risk factor for conventional APL therapy, the prognostic significance of FLT3-ITD per se remains somehow controversial.57-63 There is converging clinical evidence that arsenic therapy can overcome the poor prognosis associated with FLT3-ITD.45,47,64,65 Yet the basis for the relative ATRA resistance of FLT3-ITD APLs has not been comprehensively explored. Knock-in alleles of FLT3-ITD enhance self-renewal and drive a chronic myeloproliferative disease.66 Retroviral transduction of FLT3-ITD in PML/RARA transgenic cells accelerates APL development in mice,67 and in that setting, ATRA response is accelerated by FLT3 inhibitors.68,69 Here we reassessed the role of FLT3-ITD in APL development and therapy response. We demonstrate that FLT3-ITD impedes ATRA-induced APL differentiation and clearance in vivo. Such resistance was abrogated by combination with arsenic. This mechanistically explains the potency of the frontline ATRA/arsenic combination in these APLs.
Methods
Ethics statement
All experiments in mice were performed in accordance with a protocol approved by the Comité d’Ethique en Expérimentation Animale Paris-Nord no. 121, (project no. 02577.02).
Animal models
Female FVB/N and C57Bl/6Nj mice (6 weeks old) were purchased from Janvier Labs. Bone marrows from MRP8-PML/RARA transgenics70 (P/R) crossed with FTL3ki66 (P/R-FLT3ki) were injected in CD45.2 congenic FVB/N-strain syngenic mice. PML/RARAC212S immortalization of primary hematopoietic progenitors was performed as previously.71 Briefly, lineage-depleted (Lin−) mouse bone marrow hematopoietic cells collected from 5-fluorouracil-treated C57Bl/6Nj Pml+/+ or Pml−/− mice were infected with retroviruses obtained by transient transfection of Plat-E cells with pMSCV-PML/RARAC212S and pMIE-FLT3-ITD. After spinoculation, transduced cells were injected in C57Bl/6Nj syngenic mice. For experiments, 1 × 106 leukemic bone marrow cells were intravenously injected into recipient mice. Retinoic acid (ATRA) was administered by subcutaneous implantation of 21-day-release 10-mg pellets (Innovative Research of America). Arsenic (concentration 1 mg/mL, injected dose 5 µg/g body weight; ACROS Organics) was administered by daily intraperitoneal injection.
Retroviral transduction
Plat-E cells were transfected with retroviral vectors. Leukemic cells or Lin− progenitors were then transduced with the obtained retroviral supernatants. After spinoculation, transduced cells were injected into syngenic mice.
FACS analysis
Immunophenotypic assays were performed by first blocking cellular Fc receptors with normal rat immunoglobulin G (Fc Block, Clone 93 anti-CD16 / 32, eBioscience) and then using fluorochrome-conjugated monoclonal antibodies (1:1000): APC-conjugated anti-CD117 (Clone 2B8, eBioscience), APC-Cy7-conjugated anti-CD11b (Clone M1/70, BioLegend), and phycoerythrin-conjugated anti-Gr1 (Clone RB6-8C5, eBioscience). Staining was performed overnight at 4°C. Cells were washed and resuspended in phosphate-buffered saline (PBS) with paraformaldehyde 0.2%. Apoptosis assays were carried out according to the suppliers’ recommendations with APC-Annexin-V (BD Biosciences) and phycoerythrin-active-caspase-3 apoptosis kits (BD Biosciences). Fluorescence-activated cell sorter (FACS) analyses are performed on BD LSRFortessa Cell Analyzer (BD Biosciences).
Western immunoblotting
Total cell extract was obtained from bone marrow cells by direct lysis in Laemmli buffer. Extracts were resolved on 4% to 15% precast sodium dodecyl sulfate-polyacrylamide gel electrophoresis gels (BIO-RAD) and transferred onto nitrocellulose membranes (BioTrace NT, PALL Corp.). PML/RARA was detected with rabbit polyclonal antibodies to human PML (homemade antibody, 1:10 000), RARA with rabbit anti-RARA (C-20) polyclonal antibodies (sc-551, Santa Cruz Biotechnology, 1:1000), and vinculin with mouse monoclonal antibodies (sc-73614, Santa Cruz Biotechnology, 1:1000) as a loading control. Detection was performed with the chemiluminiscent substrate SuperSignal West Femto and Dura (Thermo Fisher Scientific).
Immunofluorescence
Next, 5 × 104 bone marrow cells were cytospinned, fixed with 10% paraformaldehyde, and permeabilized in PBS 0.2% Triton-X100 for 15 minutes. Then blocking was performed with PBS 0.05% Triton-X100 1% BSA for 30 minutes. Used primary antibodies were rabbit anti-human PML (homemade, 1:1000), Alexa Fluor A488-conjugated antimurine PML (homemade, 1:2000) for 1 hour. Rabbit secondary antibodies (1:2000) conjugated to Alexa Fluor A488 or A647 fluorochromes incubated for 30 minutes. A drop of Mowiol mounting medium containing DAPI (1 µg/mL) was finally added.
Transcriptomic analysis
Total RNA was extracted from green fluorescent protein (GFP)-sorted APL bone marrow cells (RNeasy Mini Kit, Qiagen). Transcriptomic analyses were performed at the Institut Universitaire d’Hématologie plate-form. Samples were hybridized on GeneChip Mouse Transcriptome Assay 1.0. Gene set enrichment analysis (GSEA) was performed.
Results
A FLT3-ITD knock-in allele does not accelerate PML/RARA-driven APL development
We and others had previously observed that when a FLT3-ITD was expressed from a retrovirus in PML/RARA transgenic bone marrow,67,69 activated FLT3 accelerated APL development. To further analyze the effect of the FLT3-ITD mutation on APL development, we first crossed animals bearing a FLT3-ITD knock-in allele with our MRP8-hPML/RARA transgenics. FLT3-ITD line (referred below as FLT3ki) was obtained in a C57BL/6J (B6) background, whereas hPML/RARA animals (P/R) were in the FVB/n background. Bone marrow cells from F1 P/R-FLT3ki animals were transplanted into lethally irradiated recipients. Intriguingly, there was only a trend toward earlier illness in the P/R-FLT3ki animals, with no statistically significant acceleration of leukemia development when compared with P/R alone (Figure 1A). We thus backcrossed the FLT3-ITD allele into the FVB/n background for 7 generations and generated P/R-FLT3ki animals whose bone marrow cells were transplanted into irradiated syngenic FVB/n animals. Comparing the overall and acute leukemia-free survival curves for the transplanted P/R-FLT3ki cohort along with the similar P/R cohort again showed that the P/R-FLT3ki combination did not accelerate leukemia development when compared with P/R alone (data not shown). Histopathologic findings in the P/R and P/R-FLTki leukemias showed increased immature myeloid cells occupying large areas of the bone marrow and spleen and infiltrating other organs including liver and kidney (supplemental Figure 1, available on the Blood Web site).
![Figure 1. Effect of the FLT3-ITD knock-in allele on APL initiation by PML/RARA. (A) Overall survival and acute leukemia-free survival of lethally irradiated recipients transplanted with P/R-FLT3ki, FLT3ki, or P/R bone marrow cells. (B) Strategy to obtain P/R and P/R-FLT3ki cells in a FVB background, subsequently tagged with GFP by retroviral transduction. (C) GSEA analysis for 50 hallmark gene sets between P/R and P/R-FLT3ki mice. Transcriptomic analysis performed on RNA extracted from GFP-positive leukemic cells. Only significant pathways were represented (with false discovery rate q values [FDR.q.val] <0.05) according to normalized enrichment score (NES) value.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/133/13/10.1182_blood-2018-07-866095/3/m_blood866095f1.png?Expires=1770132918&Signature=elBfaepLANSp2CwdxXPhDBfiF6DJuj4-nCOUeHOAYhxzDDu87UKdreLh1AhrZvf-umXYOEt6fguZn8OmwvfHNsO5zqyJodMMOv9FRbPboK7aob8If8y~hKof7sGblMWCUdppaK~0WmZ4OHy95rw-ecNzuzY1OUxI4HpGXogkMP4tQf6XU8iSfKPKrsIizxB9UY2r86jfG3IB3V67OqrtG-TI8skT7baqpZJ9mB5STfydHFHfgxU-R2LLPXfRvpVw2UZ6oVX7cRVYUe4PCjs2R8Hie0XcDFYTy2EBKNPmjTTR9XD~smw0JpRdCTHdaXaUXkeDc1-I8nVIJTysL9s3EQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Effect of the FLT3-ITD knock-in allele on APL initiation by PML/RARA. (A) Overall survival and acute leukemia-free survival of lethally irradiated recipients transplanted with P/R-FLT3ki, FLT3ki, or P/R bone marrow cells. (B) Strategy to obtain P/R and P/R-FLT3ki cells in a FVB background, subsequently tagged with GFP by retroviral transduction. (C) GSEA analysis for 50 hallmark gene sets between P/R and P/R-FLT3ki mice. Transcriptomic analysis performed on RNA extracted from GFP-positive leukemic cells. Only significant pathways were represented (with false discovery rate q values [FDR.q.val] <0.05) according to normalized enrichment score (NES) value.
Effect of the FLT3-ITD knock-in allele on APL initiation by PML/RARA. (A) Overall survival and acute leukemia-free survival of lethally irradiated recipients transplanted with P/R-FLT3ki, FLT3ki, or P/R bone marrow cells. (B) Strategy to obtain P/R and P/R-FLT3ki cells in a FVB background, subsequently tagged with GFP by retroviral transduction. (C) GSEA analysis for 50 hallmark gene sets between P/R and P/R-FLT3ki mice. Transcriptomic analysis performed on RNA extracted from GFP-positive leukemic cells. Only significant pathways were represented (with false discovery rate q values [FDR.q.val] <0.05) according to normalized enrichment score (NES) value.
To compare the leukemic cells from these P/R and the P/R-FLT3ki models, we retrovirally transduced leukemia blasts (from leukemias that arose in the predominantly FVB/n background) with GFP, transferred these cells into FVB/n mice (Figure 1B), sorted GFP-positive cells from the marrows of resultant leukemic animals, and compared the transcriptomes of these leukemias. As expected, P/R-FLT3ki blasts exhibited activated STAT5, K-RAS, or tumor necrosis factor/NF-kB signaling, as determined by GSEA analysis.72-77 Conversely, interferon α or γ pathways were strongly downregulated in the FLT3-ITD context78 (Figure 1C). Differentiation statuses, as assessed by May-Grünwald-Giemsa (MGG) stains and cell surface analyses (Kit, CD11b, GR1), were similar (Figure 2A). Thus, although this model exhibited the expected changes downstream of FLT3 signaling, it did not lead to a sharp acceleration of APL development.67-69
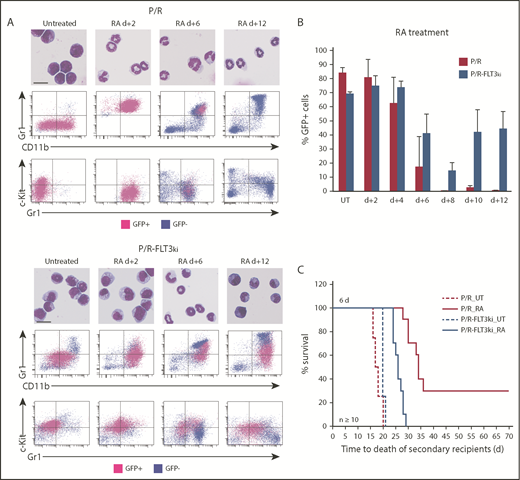
Cell differentiation and blast clearance in P/R and P/R-FLT3ki APL on ATRA treatment. (A) ATRA-induced differentiation of P/R (top) and P/R-FLT3ki (bottom) bone marrow cells, as assessed by MGG staining (scale bar, 10 µm). FACS analysis after 2, 6, and 12 days of in vivo ATRA treatment. GFP-positive leukemic cells (pink) and GFP-negative normal cells (blue) are shown. (B) Percentages of GFP-positive APL cells in bone marrow of untreated (UT) and ATRA-treated P/R (red bars) and P/R-FLT3ki (blue bars) mice at 2 to 12 days posttreatment. Data are expressed as mean ± standard deviation of 2 independent experiments. (C) Survival of secondary recipients transplanted with total APL bone marrow cells from ATRA 6-day-treated primary APL mice in both models (n ≥ 10 for each model).
Cell differentiation and blast clearance in P/R and P/R-FLT3ki APL on ATRA treatment. (A) ATRA-induced differentiation of P/R (top) and P/R-FLT3ki (bottom) bone marrow cells, as assessed by MGG staining (scale bar, 10 µm). FACS analysis after 2, 6, and 12 days of in vivo ATRA treatment. GFP-positive leukemic cells (pink) and GFP-negative normal cells (blue) are shown. (B) Percentages of GFP-positive APL cells in bone marrow of untreated (UT) and ATRA-treated P/R (red bars) and P/R-FLT3ki (blue bars) mice at 2 to 12 days posttreatment. Data are expressed as mean ± standard deviation of 2 independent experiments. (C) Survival of secondary recipients transplanted with total APL bone marrow cells from ATRA 6-day-treated primary APL mice in both models (n ≥ 10 for each model).
FLT3-ITD blunts APL cell differentiation and blast clearance on ATRA treatment
In P/R APLs, ATRA initiated terminal granulocytic differentiation assessed by MGG staining and FACS analysis (Gr1 and Mac/CD11b), already evident after 2 days.32,79 In sharp contrast, P/R-FLT3ki APLs only displayed clear features of differentiation at day 6 (Figure 2A-B). Moreover, loss of c-kit/CD117, a marker of leukemic cell self-renewal, occurred at day 2 in P/R mice, whereas c-kit expression persisted at day 6 in P/R-FLT3ki (Figure 2A-B). Examining APL clearance by GFP, we observed that ATRA cleared P/R APLs at day 8, with normal bone marrow replacing leukemia as soon as day 6.32,79 In keeping with their delayed differentiation, P/R-FLT3ki APL cell elimination was hindered at day 6 (Figure 2A-B). Moreover, ATRA treatment never eliminated P/R-FLT3ki APL cells, which actually aggressively regrew at day 10 (Figure 2B-C). As expected, loss of APL-initiating activity at day 6 was impeded in the FLT3-ITD model (Figure 2D). Altogether, FLT3-ITD mutation blocks therapeutic response to ATRA in APL mice.
FLT3-ITD impedes PML/RARA degradation and NB-reformation, not transcriptional control
To explore the basis for ATRA resistance conferred by FLT3ki expression, we first performed transcriptomic analyses at 9 and 24 hours to explore PML/RARA-mediated gene activation. Unexpectedly, robust activation of many canonical PML/RARA-target genes was observed (such as Pram1 and Cyp26a1), implying that ATRA-initiated transcription was unaffected (data not shown). Accordingly, pathway analysis showed that very similar signaling networks were activated early on in the 2 models (Figure 3A).
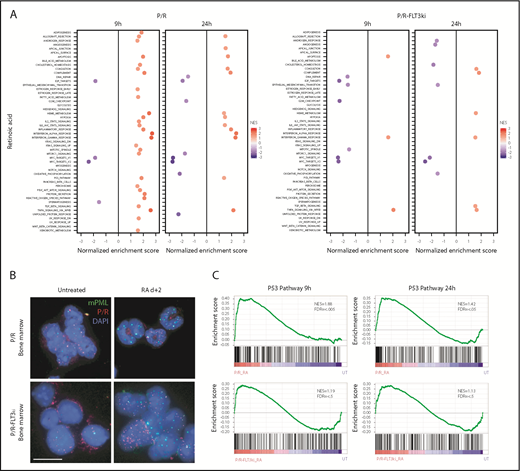
Transcriptional analysis and PML NB reformation on ATRA treatment. (A) GSEA analysis for 50 hallmark gene sets in P/R and P/R-FLT3ki mice treated or not during 9 or 24 hours with ATRA. Only significant pathways were represented (with FDR.q.val <0.05) according to NES value. (B) PML NB reformation assessed by immunofluorescence analysis of mPML (green) and human PML/RARA (red) with DAPI (blue) in bone marrow cells of P/R and P/R-FLT3ki APL mice treated with ATRA after 2 days (scale bar, 10 µm). (C) GSEA analysis of the p53 pathway in P/R and P/R-FLT3ki mice treated during 9 or 24 hours with ATRA. NES and FDR are noted. FDR.q.val <0.05 are in bold.
Transcriptional analysis and PML NB reformation on ATRA treatment. (A) GSEA analysis for 50 hallmark gene sets in P/R and P/R-FLT3ki mice treated or not during 9 or 24 hours with ATRA. Only significant pathways were represented (with FDR.q.val <0.05) according to NES value. (B) PML NB reformation assessed by immunofluorescence analysis of mPML (green) and human PML/RARA (red) with DAPI (blue) in bone marrow cells of P/R and P/R-FLT3ki APL mice treated with ATRA after 2 days (scale bar, 10 µm). (C) GSEA analysis of the p53 pathway in P/R and P/R-FLT3ki mice treated during 9 or 24 hours with ATRA. NES and FDR are noted. FDR.q.val <0.05 are in bold.
Previous studies have stressed the role of PML NB reformation in APL regression.32,36,37,80 We thus compared NB status on ATRA treatments in vivo in our 2 models. In both untreated P/R and P/R-FLT3ki APLs, human PML/RARA and murine PML (mPML) proteins appeared as microspeckles (Figure 3B).23,25 In P/R-FLT3ki APLs, PML/RARA was often detected in the cytoplasm, as described in some patients with APL.23 After 2 days of ATRA treatment, mPML was reorganized into normal-looking NB in P/R cells. In sharp contrast, this reorganization was absent from P/R-FLT3ki APLs, where microspeckles remained apparent (Figure 3B). Failure of NB reorganization might underlie ATRA resistance, and previous studies demonstrated that p53 is a key downstream effector of PML NB.36 Indeed, the network of p53 targets was activated in P/R APLs, but not in P/R-FLT3ki ones (Figure 3B-C).
We formerly demonstrated that ATRA-induced NB-reorganization was a result of PML/RARA degradation, releasing the ability of normal PML to reform NBs.81 We thus compared PML/RARA degradation in our 2 APL models in vivo. As expected, ATRA initiated PML/RARA degradation in P/R APLs (Figure 4A). In contrast, in P/R-FLT3ki APLs, PML/RARA expression was maintained by both western blot and immunofluorescence at least up to day 6 (Figure 4A-B). Inefficient PML/RARA degradation in P/R-FLT3ki APLs is the most likely origin of therapy resistance. To investigate the basis for ATRA inability to clear PML/RARA, we examined the status of the normal RARA protein, also targeted by ATRA-initiated proteolysis.81 Although RARA was rapidly downregulated in P/R APL cells, it was not in P/R-FLT3ki ones (Figure 4C).
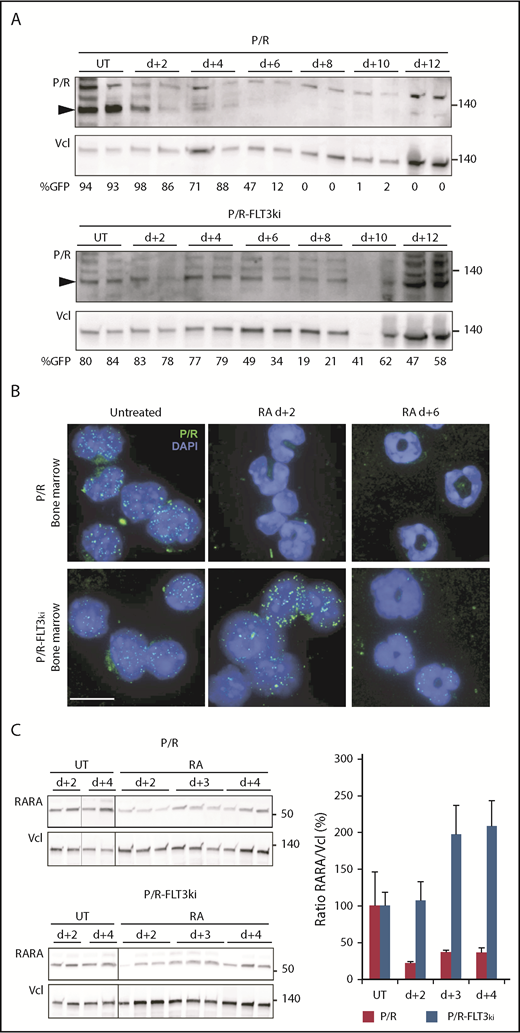
Effect of FLT3-ITD on PML/RARA degradation on ATRA treatment. (A) Western blots of hPML/RARA (P/R, arrowheaded) and vinculin (Vcl) expression in the bone marrow of P/R (top) and P/R-FLT3ki (bottom) APL untreated (UT) and ATRA-treated mice from 2 to 12 days. Antihuman PML antibody. The percentage of GFP-positive cells is indicated for each sample. (B) Immunofluorescence analysis of human PML/RARA (green, P/R) with DAPI (blue) in bone marrow cells of P/R and P/R-FLT3ki APL mice treated with ATRA after 2 and 6 days (scale bar, 10 µm). (C) Western blot analysis of RARA degradation on in vivo ATRA treatment of the indicated time in both APL models (left). Graph shows quantification of RARA/Vcl ratio, indicative of RARA degradation (right).
Effect of FLT3-ITD on PML/RARA degradation on ATRA treatment. (A) Western blots of hPML/RARA (P/R, arrowheaded) and vinculin (Vcl) expression in the bone marrow of P/R (top) and P/R-FLT3ki (bottom) APL untreated (UT) and ATRA-treated mice from 2 to 12 days. Antihuman PML antibody. The percentage of GFP-positive cells is indicated for each sample. (B) Immunofluorescence analysis of human PML/RARA (green, P/R) with DAPI (blue) in bone marrow cells of P/R and P/R-FLT3ki APL mice treated with ATRA after 2 and 6 days (scale bar, 10 µm). (C) Western blot analysis of RARA degradation on in vivo ATRA treatment of the indicated time in both APL models (left). Graph shows quantification of RARA/Vcl ratio, indicative of RARA degradation (right).
Combined RA/As treatment overcomes FLT3-ITD-triggered resistance
Multiple clinical studies have now demonstrated the potency of the ATRA/arsenic combination, including in FLT3-mutant APLs.44,45,64,65,79,82 We thus examined the effects of the ATRA/arsenic combination in our 2 APL models. In keeping with previous studies reporting antagonism between ATRA and arsenic for differentiation,32,34 a slight delay in ATRA-initiated blast maturation was observed in P/R treated with the combination (Figures 2A and 5A). Remarkably, as early as day 2, massive cellular differentiation was now observed in both APL models treated with the combination (Figure 5A). Terminal differentiation was followed by rapid APL clearance, as determined by GFP positivity in the bone marrow, and restoration of normal hematopoiesis at day 6 in P/R-FLT3ki APLs (Figure 5B). Rapid loss of Kit, a key marker of self-renewal, may explain why addition of arsenic-promoted clearance of P/R-FLT3ki APLs. We also assessed apoptosis induction using Annexin-V and active Caspase-3 labeling (Figure 5C). Remarkably, P/R-FLT3ki APL did not exhibit enhanced apoptosis on ATRA therapy, whereas both P/R-FLT3ki and P/R APLs displayed clear apoptosis induction when treated with the arsenic/ATRA combination (Figure 5C). As expected from the nonoverlapping degrons,83 PML/RARA degradation was accelerated by the combined treatment in P/R-FLT3ki mice (Figure 5D). Adding arsenic to ATRA allowed very early restoration of mPML NBs at day 2, with a similar pattern in the 2 APL models (Figure 5E).
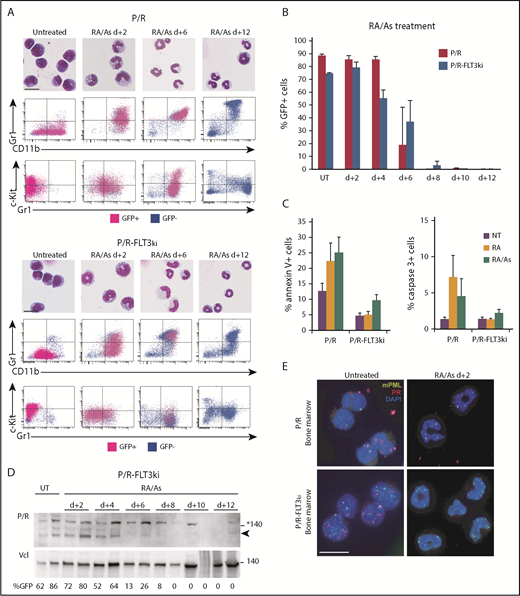
Arsenic drives elimination of FLT3ki APLs. (A) ATRA/As-induced differentiation of P/R (top) and P/R-FLT3ki (bottom) bone marrow cells, as assessed by MGG staining (scale bar 10, µm) and FACS analysis after 2, 6, 8, and 12 days of in vivo treatment. GFP-positive leukemic cells (pink) and GFP-negative normal cells (blue) are shown. (B) Percentages of GFP-positive APL cells in bone marrow of untreated (UT) and ATRA/As-treated P/R (red bars) and P/R-FLT3ki (blue bars) mice at 2 to 12 days posttreatment. (C) Proportion of annexin-V (left) and active caspase-3 (right)-positive spleen cells in APL mice treated for 4 days, as indicated. (D) Western blot of PML/RARA (P/R, arrowheaded) and vinculin (Vcl) expression in the bone marrow of P/R-FLT3ki APL UT and ATRA/As-treated mice from 2 to 12 days. (E) PML NB reformation assessed by immunofluorescence analysis of mPML (green) and human PML/RARA (red) with 4′,6-diamidino-2-phenylindole (DAPI) (blue) in bone marrow cells of P/R and P/R-FLT3ki APL mice treated with ATRA/As after 2 days (scale bar, 10 µm).
Arsenic drives elimination of FLT3ki APLs. (A) ATRA/As-induced differentiation of P/R (top) and P/R-FLT3ki (bottom) bone marrow cells, as assessed by MGG staining (scale bar 10, µm) and FACS analysis after 2, 6, 8, and 12 days of in vivo treatment. GFP-positive leukemic cells (pink) and GFP-negative normal cells (blue) are shown. (B) Percentages of GFP-positive APL cells in bone marrow of untreated (UT) and ATRA/As-treated P/R (red bars) and P/R-FLT3ki (blue bars) mice at 2 to 12 days posttreatment. (C) Proportion of annexin-V (left) and active caspase-3 (right)-positive spleen cells in APL mice treated for 4 days, as indicated. (D) Western blot of PML/RARA (P/R, arrowheaded) and vinculin (Vcl) expression in the bone marrow of P/R-FLT3ki APL UT and ATRA/As-treated mice from 2 to 12 days. (E) PML NB reformation assessed by immunofluorescence analysis of mPML (green) and human PML/RARA (red) with 4′,6-diamidino-2-phenylindole (DAPI) (blue) in bone marrow cells of P/R and P/R-FLT3ki APL mice treated with ATRA/As after 2 days (scale bar, 10 µm).
To investigate transcriptional signaling on dual ATRA/arsenic treatment in P/R and P/R-FLT3ki APLs, we profiled GFP-sorted cells 9 and 24 hours after in vivo treatment. As before, the 2 models showed essentially similar signaling pathways modulation (Figure 6A). Yet, critically, arsenic rescued the defective activation of p53 signaling in P/R-FLT3ki APLs at 9 hours (Figure 6A-B). Accordingly, the ATRA/arsenic combination strongly impeded APL transplantability (Figure 6C compared to Figure 2C). Collectively, these data establish that resistance conferred by FLT3-ITD can be fully reversed by the addition of arsenic to ATRA treatment.
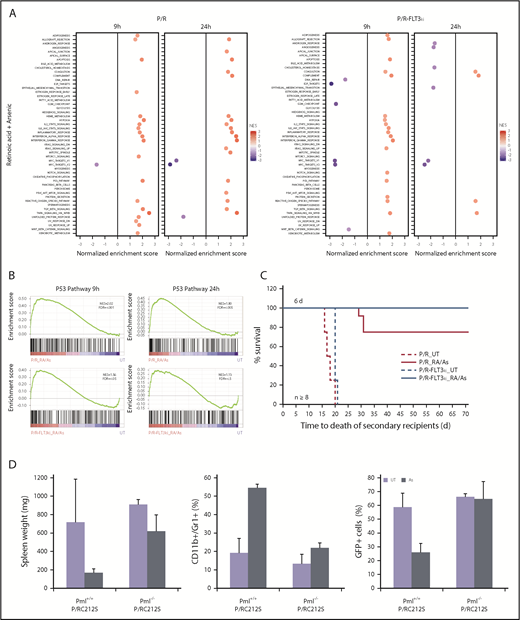
Combined ATRA/As treatment overcomes FLT3-ITD-triggered resistance. (A) GSEA analysis for 50 hallmark gene sets between P/R and P/R-FLT3ki mice treated during 9 or 24 hours with ATRA/As. Only significant pathways were represented (with FDR.q.val <0.05) according to NES value. (B) GSEA analysis of the p53 pathway in P/R and P/R-FLT3ki mice treated during 9 or 24 hours with ATRA/As. NES and FDR are noted. FDR.q.val <0.05 are in bold. (C) Survival of secondary recipients transplanted with APL bone marrow cells from ATRA/As 6-day-treated primary APL mice in both models (n ≥ 8 for each model). (D) Comparison of spleen weight (left), differentiation (CD11+/Gr1+, middle) and blast clearance (GFP+, right) of untreated (UT) or As 7-day-treated APL mice with MRP8-PML/RARAC212S in Pml+/+ or Pml−/− genetic background. Data are expressed as mean ± standard deviation of at least 2 independent experiments.
Combined ATRA/As treatment overcomes FLT3-ITD-triggered resistance. (A) GSEA analysis for 50 hallmark gene sets between P/R and P/R-FLT3ki mice treated during 9 or 24 hours with ATRA/As. Only significant pathways were represented (with FDR.q.val <0.05) according to NES value. (B) GSEA analysis of the p53 pathway in P/R and P/R-FLT3ki mice treated during 9 or 24 hours with ATRA/As. NES and FDR are noted. FDR.q.val <0.05 are in bold. (C) Survival of secondary recipients transplanted with APL bone marrow cells from ATRA/As 6-day-treated primary APL mice in both models (n ≥ 8 for each model). (D) Comparison of spleen weight (left), differentiation (CD11+/Gr1+, middle) and blast clearance (GFP+, right) of untreated (UT) or As 7-day-treated APL mice with MRP8-PML/RARAC212S in Pml+/+ or Pml−/− genetic background. Data are expressed as mean ± standard deviation of at least 2 independent experiments.
Arsenic targets both hPML/RARA and mPML, possibly explaining its potency, even with the relative ATRA resistance conferred by FLT3 activation. To decipher the contribution of each partner, we generated APL models by infection of bone marrow progenitors with retroviruses encoding FLT3-ITD and PML/RARAC212S, a protein that is defective for arsenic binding.84 We also assessed arsenic response in APLs derived from Pml+/+ or Pml−/− progenitors.85 Using both spleen weight and GFP-positive cells as end points, Pml−/− APLs were completely resistant to arsenic when compared with Pml+/+ ones, stressing the role of normal PML in therapy response (supplemental Figure 2). Unexpectedly, APLs expressing PML/RARAC212S remained partially sensitive to arsenic (Figure 6D), demonstrating the key role of arsenic targeting of normal PML protein in therapy response.37,80,86
Discussion
Previous studies found that FLT3 cooperated with PML/RARA to accelerate APL development,67 and that FLT3 inhibitors synergized with ATRA to initiate differentiation in these models. Yet these studies did not explore/report ATRA response.69 Our experiments demonstrate that a knock-in FLT3-ITD allele does not accelerate APL development, but dramatically deters ATRA response. The reasons underlying these discrepancies may relate to higher levels of FLT3-ITD expression on retroviral expression in the previous studies. Yet most of our results were duplicated in a distinct model of FLT3-ITD APL mice, where the FLT3-ITD gene was directly transduced in P/R cells (supplemental Figure 3). Our observations shed a new light on multiple clinical observations that this common alteration is associated with an increased probability of relapses with ATRA/chemotherapy regimen. Interestingly, amplitude of ex vivo differentiation was linked to survival with suboptimal ATRA/chemotherapy associations, a finding that could now be interpreted as the possible effect of FLT3-ITD on ATRA-induced ex vivo differentiation.87 Mechanistically, FLT3 activation impedes ATRA-induced PML/RARA and RARA degradation. This consequently inhibits PML NB reformation and p53 activation, strongly supporting our model for a key role of this cascade in APL therapy by ATRA.36,37 Previous studies demonstrated multiple cross talks between ATRA or RARA and kinases activated downstream of FLT3.88,89 Biochemical attempts to involve them in inefficient ATRA-induced PML/RARA degradation were not successful. It is also formally possible that the delayed PML/RARA degradation also reflects FLT3-ITD-enhanced APL cell self-renewal, constantly restoring a new pool of PML/RARA-expressing cells. In that respect, STAT5, a key pathway of AML self-renewal, was clearly activated by FLT3-ITD (Figure 1C).90
We unambiguously demonstrate that ATRA resistance in P/R-FLT3ki APLs can be circumvented by arsenic, which restores PML/RARA degradation, NB-reformation, p53 activation, and loss of self-renewal in vivo. This likely reflects the ability of arsenic to target PML/RARA for destruction by biochemical pathways distinct from ATRA-activated ones. Yet arsenic also directly targets normal PML for NB reformation. Critically, we provide evidence strongly supporting implication of normal PML in murine APL arsenic response. Not only are APLs obtained in a Pml−/− background completely arsenic-resistant, but transduction of the arsenic-resistant PML/RARAC212S mutant84 in Pml+/+ background nevertheless exhibits some response to arsenic in vivo. Indeed, in P/R-FLT3ki APLs treated with the ATRA/arsenic combination, NB reformation was more rapidly restored than PML/RARA degradation (Figure 5D-E). Note that mutations in the arsenic binding site of normal PML were observed in therapy-resistant patients,52,53,80,86 pointing to the human relevance of our observations. Clinical evidence suggested that addition of arsenic to APL therapy (either frontline or later in treatment) can erase the poor prognosis associated with FLT3 activation and/or hyperleukocytosis.45,47,64,65,91,92 Our studies imply that first-line arsenic should be used in FLT3-ITD or high-risk patients (in combination or not with some chemotherapy). More broadly, our findings could point to possible deregulation of basal ATRA/RARA signaling in tumors with activated kinase signaling.89,93,94 Our findings also provide some evidence for a role of arsenic in targeting the normal PML allele independently from PML/RARA, raising the possibility that arsenic may be active through PML targeting in selected conditions, even where PML is not rearranged.95
All renewable reagents are available on request to the corresponding author. Transcriptomic data are publicly available at European Bioinformatics Institute, Array Express E-MTAB-7566.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank N. Setterblad and Institut Universitaire d’Hématologie technology platforms for imaging, fluorescence-activated cell sorter analysis, and transcriptomic assays; V. Parietti and M. Chopin for the animal house; and laboratory members for critical reading of the manuscript.
The H.d.T. laboratory is supported by the Ligue Nationale contre le Cancer, INSERM, Centre National de la Recherche Scientifique, University Paris Diderot, Institut National du Cancer, the Association pour la Recherche contre le Cancer (Prix Griffuel), European Research Council Senior grant 268729-STEMAPL (to H.d.T.), and TRANSCAN (DRAMA project). The contributions of C.G. and S.K. were supported by National Institutes of Health, National Cancer Institute grant R01-CA95274.
Authorship
Contribution: C.E, R.R., and K.L.R. performed and analyzed in vivo experiments on P/R and P/R-FLT3ki mouse models; C.B. and A.-L.M. performed in vivo experiments on PML/RARAC212S in Pml+/+ and Pml−/− mice; C.G. generated and characterized P/R-FLT3ki mice; S.Q. performed the bioinformatics analysis; S.K. provided P/R-FLT3ki murine bone marrows; C.E, R.R., S.K, and H.d.T reviewed the data and wrote the manuscript with the assistance and final approval of all authors; and H.d.T. designed the study.
Conflict-of-interest disclosure: H de Thé consults for Vectorlabs Gmbh. The remaining authors declare no competing financial interests.
The current affiliation for K.L.R. is Phylogica, Subiaco, WA, Australia.
The current affiliation for C.G. is Genentech Inc, South San Francisco, CA.
Correspondence: Hugues de Thé, INSERM 944 Hôpital St. Louis 1 Ave Vellefaux, Paris, 75475 France; e-mail: hugues.dethe@inserm.fr.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal