

Abstract
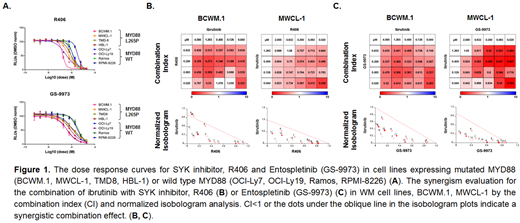
Activating mutations in MYD88, a component of the Toll-receptor (TLR) pathway, trigger Myddosome self-assembly and multiple downstream pro-survival signaling through BTK and IRAK4/IRAK1 (Yang et al, Blood 2013). Activation of B-cell receptor (BCR) signaling has also been observed in WM, though the mechanism(s) remain to be clarified (Argyropoulos et al, Leukemia 2016). While activating mutations in the BCR components CD79A/B are common in MYD88 mutated ABC DLBCL, they are uncommon in WM (Ngo et al, Nature 2011; Hunter et al, Blood 2014). We therefore sought to clarify if TLR/MYD88 crosstalk could explain aberrant BCR signaling in WM. We performed PhosFlow analysis of MYD88 and BCR signaling components of MYD88 mutated and wild-type WM and ABC DLBCL cell lines. These studies showed high levels for expression of the BCR component p-SYK (Y525-526) in MYD88 mutated WM (BCWM.1, MWCL-1) and ABC DLBCL (TMD-8, HBL-1) cell lines versus MYD88 wild-type cell lines. High levels of p-SYK were also observed in primary MYD88 mutated WM cells compared to healthy donor peripheral blood B-cells. Following treatment with a MYD88 blocking peptide, p-SYK was robustly reduced in WM cell lines, while only modest reduction was observed in ABC DLBCL cell lines which also carry activating CD79B mutations. Use of MYD88 inhibitor also blocked the p-SYK in primary MYD88 mutated WM cells. Lentiviral over-expression of MYD88 L265P but not wild-type MYD88 or vector control augmented p-SYK expression in MYD88 mutated BCWM.1 WM cells, as well as MYD88 wild-type Ramos and OCI-Ly7 cells. Conversely, knockdown of MYD88 in BCWM.1 cells confirmed the dependence of p-SYK on mutated MYD88. p-SYK was also confirmed to be in complex with the Myddosome in BCWM.1 cells by co-immunoprecipitation (co-IP) using anti-MYD88 and anti-SYK/anti-p-SYK specific antibodies. Over-expression and knockdown studies of mutated MYD88 also showed an association of downstream p-STAT3 (Y705) signaling on MYD88 triggered p-SYK, a finding confirmed by use of SYK inhibitors which blocked p-STAT3 in a dose-dependent manner. CellTiter-Glo® viability assays showed the SYK inhibitors, R406 and Entospletinib (GS-9973), produced higher cytotoxicity in MYD88 mutated versus wild-type B-cell lines. (Figure 1A) Since BCR/SYK kinase triggers divergent signaling from TLR/BTK/IRAK pathways, we examined the combined effect of BTK and SYK inhibition. Combination index and normalized isobologram analysis demonstrated synergistic effects with the combination of ibrutinib with either R406 or Entospletinib in MYD88 mutated WM and ABC DLBCL cell lines (Figure 1B, 1C). The combination of ibrutinib with either R406 or Entospletinib also produced more robust tumor cell apoptosis in primary MYD88 mutated WM cells. Our findings show that MYD88 can cross-talk with BCR pathway through SYK tyrosine kinase, and trigger aberrant p-STAT3 signaling. The inhibition of both TLR/BTK and BCR/SYK activation by use of ibrutinib and SYK inhibitors produced synergistic tumor cell killing of MYD88 mutated WM and ABC DLBCL lymphomas, and provides a framework for clinical studies aimed at extinguishing aberrant MYD88 signaling.
Castillo:Millennium: Research Funding; Pharmacyclics: Consultancy, Research Funding; Janssen: Consultancy, Research Funding; Genentech: Consultancy; Beigene: Consultancy, Research Funding; Abbvie: Consultancy, Research Funding. Hunter:Pharmacyclics: Consultancy. Gray:Syros, Soltego, Petra, C4 Therapeutics: Equity Ownership. Treon:Pharmacyclics: Consultancy, Other: Travel, Accommodations, Expenses, Research Funding; BMS: Research Funding; Johnson & Johnson: Consultancy; Janssen: Consultancy, Other: Travel, Accommodations, Expenses.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal