

TO THE EDITOR:
We have identified 9 affected individuals from 3 unrelated families with macrothrombocytopenia and mild to moderate bleeding diathesis inherited due to missense mutations in the glucosamine (UDP-N-acetyl)-2-epimerase/N-acetylmannosamine kinase (GNE) gene. Previous reports have primarily described a myopathy associated with GNE variants. Eight of the 9 patients in our cohort had no evidence of myopathy, and the 1 patient with neuromuscular symptoms had a muscle biopsy inconsistent with GNE myopathy. Following is the description of our patients and the variants identified as well as the laboratory evidence supporting pathogenicity for the identified variants and describing our hypothesized mechanism for thrombocytopenia.
Platelets express the negatively charged, N-acetylneuraminic acid–containing pentasaccharide sialic acid, most of which resides on surface glycoprotein carbohydrate side chains.1 A large proportion of the platelet sialic acid is neuraminidase labile and is implicated in platelet functional events, such as aggregation and adhesion, as well as contributing to the platelet's electrophoretic mobility.2 Sialylation appears to affect the number of circulating platelets. Removal of sialic acid (desialylation) shortens platelet life span3 and triggers clearance of senescent platelets.4
Previous data in both human and animal models suggest a role for platelet sialylation in regulation of platelet count5-7 and hint that genes that alter platelet glycoprotein expression may result in differences in baseline platelet numbers. In contrast, most previous reports of patients with GNE variants have described primarily a myopathy, although some patients with associated mild, asymptomatic thrombocytopenia have been described in 0% to 2.5% of GNE myopathy registry patients (Argov Avisohar, Hadassah-Hebrew University Medical Center, e-mail, 2017).8,9 GNE regulates and initiates biosynthesis of N-acetylneuraminic acid, a precursor of sialic acids, which is required for normal sialylation in hematopoietic cells.10
The medical records of 9 patients from 3 distinct families (F1, F2, and F3), presenting with bleeding and macrothrombocytopenia, and found to have GNE gene variants, were reviewed. Detailed description of genetic analysis and other methods is given in the supplemental materials, available on the Blood Web site. Data concerning clinical presentation, platelet numbers and morphology, muscle weakness, and genetic findings were collected. All participants provided consent according to local institutional policies, including consent for DNA analysis.
F1 and F2 are of Palestinian Arab descent with 5 and 3 affected individuals (P1-P5 and P6-P8). F1 cases had a novel variant in GNE (c.1516_1517delinsTT, p.Gly475Phe) (supplemental Figure 2). In F2, a homozygous variant in GNE (c.1457T>C, p.Leu486Pro) was found (supplemental Figure 2). Both variants in F1 and F2 occur in a highly conserved region of exon 9. F3 involves a 13-year-old male Caucasian (P9) born to nonconsanguineous parents of mixed European lineage (Figure 1A; Table 1). In F3, analysis revealed 2 rare variants: (c.1649A>G, p.Asn550Ser) previously reported in myopathy11 and c.562C>T, p.His188Tyr occurring in a highly conserved region in exon 4 (supplemental Figure 2). All 4 variants were unobserved in several databases of healthy individuals. Clinically, all affected family members report bruising ± epistaxis. Affected women also reported severe menorrhagia and recurrent episodes of hemorrhagic corpus luteum requiring red cell and platelet transfusions. Three affected individuals had a history of surgery or trauma requiring red cell and platelet transfusions. Importantly, in all but 1 patient, there was no evidence of clinical myopathy (P1-P8), with exclusion of subclinical myopathy in 2 family members (P1 and P4) by magnetic resonance imaging.12 P9 has hypotonia of the lower extremities, as well as autism spectrum disorder.13 Muscle biopsy was not consistent with GNE myopathy (supplemental Figure 1).
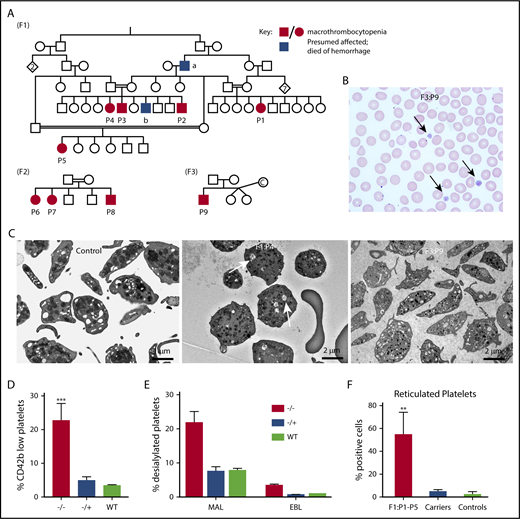
Family and biology studies supporting GNE pathobiology.(A) Family pedigrees. Each family is represented by the designation F1-3. Patients P1 through P9 are shown as black circles (female) and squares (males), whereas unaffected family members are shown as open symbols. Blue boxes refer to potentially affected individuals, but no blood counts were done. Red boxes/circles represent affected individuals. F1 pedigree: The common ancestor “a” died of posttrauma bleeding, whereas “b” died at the age of 2 years from an intracranial hemorrhage. Parents of patients have normal platelet counts and morphology. F2 pedigree, F3 pedigree: “c” refers to an egg donor. (B) A representative hematoxylin and eosin peripheral smear from a patient. Arrows indicate large platelets. (Original magnification, ×60; hematoxylin and eosin stain). (C) Transmission electron microscopy images of platelets isolated from control and indicated patients. Likely dilated open canalicular membrane system that engulfed some debris is shown by the arrows. Bar shown indicates image size. (D) Comparison of percentage of platelets (by FSC/SSC and expression of CD42) that are CD42b low expressing in patients vs heterozygous carriers and unaffected controls. n = 5 patients, 4 heterozygous family members, and 4 unrelated controls. **P = .002 vs control. (E) Fluorescence-activated cell sorter analysis of platelet sialylation levels demonstrating loss of sialylation in platelets from patients with GNE variants. ***P < .001 vs control for MAL. (F) Fluorescence-activated cell sorter analysis of total platelet population on F1 patients (P1 through P5), an obligate F1 carrier (father of P2-P4), and 3 nonaffected controls showing staining for thiazole orange. For panels D and F, graphs show mean ± 1 standard deviation. ***P < .02 comparing affected patients with control patients by Student t test. MAL, maackai amurensis lectin II; SNA/EBL, sambucus nigra lectin; WT, wild type.
Family and biology studies supporting GNE pathobiology.(A) Family pedigrees. Each family is represented by the designation F1-3. Patients P1 through P9 are shown as black circles (female) and squares (males), whereas unaffected family members are shown as open symbols. Blue boxes refer to potentially affected individuals, but no blood counts were done. Red boxes/circles represent affected individuals. F1 pedigree: The common ancestor “a” died of posttrauma bleeding, whereas “b” died at the age of 2 years from an intracranial hemorrhage. Parents of patients have normal platelet counts and morphology. F2 pedigree, F3 pedigree: “c” refers to an egg donor. (B) A representative hematoxylin and eosin peripheral smear from a patient. Arrows indicate large platelets. (Original magnification, ×60; hematoxylin and eosin stain). (C) Transmission electron microscopy images of platelets isolated from control and indicated patients. Likely dilated open canalicular membrane system that engulfed some debris is shown by the arrows. Bar shown indicates image size. (D) Comparison of percentage of platelets (by FSC/SSC and expression of CD42) that are CD42b low expressing in patients vs heterozygous carriers and unaffected controls. n = 5 patients, 4 heterozygous family members, and 4 unrelated controls. **P = .002 vs control. (E) Fluorescence-activated cell sorter analysis of platelet sialylation levels demonstrating loss of sialylation in platelets from patients with GNE variants. ***P < .001 vs control for MAL. (F) Fluorescence-activated cell sorter analysis of total platelet population on F1 patients (P1 through P5), an obligate F1 carrier (father of P2-P4), and 3 nonaffected controls showing staining for thiazole orange. For panels D and F, graphs show mean ± 1 standard deviation. ***P < .02 comparing affected patients with control patients by Student t test. MAL, maackai amurensis lectin II; SNA/EBL, sambucus nigra lectin; WT, wild type.
Clinical and laboratory characteristics of patients in the 3 families
| F1 | F2 | F3 | |
|---|---|---|---|
| Number of patients | 3 F, 2 M (P1-P5); | 2 F, 1 M (P6-P8); | 1 M (P9); |
| Age, y | 24-42 | 6-14 | 11 |
| Bleeding symptoms | Easy bruising, epistaxis, menorrhagia, hemorrhagic corpus luteum | Easy bruising, menorrhagia | Easy bruising, epistaxis |
| Platelet count (N 150-450 × 109/L) | 1-4 × 109/L | 3-10 × 109/L | 30-40 × 109/L |
| MPV | NA | NA | 18.9-19.4 fL |
| Hemoglobin (N 12-16 g/dL) | 10.7-14.4 g/dL | 9.37 g/dL* | Normal |
| Blood smear | Macrothrombocytopenia; no pale platelets | Macrothrombocytopenia; no pale platelets | Macrothrombocytopenia; no pale platelets |
| Bone marrow | Not done | Mild increase in megakaryocytes with hypolobulated forms and focal mild increase reticulin fibers* | Not done |
| RBC transfusions | Yes | Yes | No |
| Platelet transfusions | Yes | Yes | Yes |
| Myopathy | No | No | Yes |
| F1 | F2 | F3 | |
|---|---|---|---|
| Number of patients | 3 F, 2 M (P1-P5); | 2 F, 1 M (P6-P8); | 1 M (P9); |
| Age, y | 24-42 | 6-14 | 11 |
| Bleeding symptoms | Easy bruising, epistaxis, menorrhagia, hemorrhagic corpus luteum | Easy bruising, menorrhagia | Easy bruising, epistaxis |
| Platelet count (N 150-450 × 109/L) | 1-4 × 109/L | 3-10 × 109/L | 30-40 × 109/L |
| MPV | NA | NA | 18.9-19.4 fL |
| Hemoglobin (N 12-16 g/dL) | 10.7-14.4 g/dL | 9.37 g/dL* | Normal |
| Blood smear | Macrothrombocytopenia; no pale platelets | Macrothrombocytopenia; no pale platelets | Macrothrombocytopenia; no pale platelets |
| Bone marrow | Not done | Mild increase in megakaryocytes with hypolobulated forms and focal mild increase reticulin fibers* | Not done |
| RBC transfusions | Yes | Yes | No |
| Platelet transfusions | Yes | Yes | Yes |
| Myopathy | No | No | Yes |
P1 though P9 refers to patient designation in Figure 1.
F, female; M, male; NA, not available; RBC, red blood cell.
Available only for P7. Iron deficiency anemia was noted only in female patients secondary to menorrhagia.
The presence of macrothrombocytopenia was confirmed on light microscopy (Figure 1B). Electron microscopy supported the presence of large platelets (Figure 1C). By flow cytometry, patient platelets were large and much more heterogeneous by forward and side scatter than control platelets. Costaining platelets for CD42a (GPIX) and its relative level to CD42b (GPIb) allow an estimate of degree of cleavage of the extracellular glycocalicin domain of GPIbα.4 Control platelets showed little to no cleavage, whereas F1 and F3 patient platelets demonstrated a population of platelets with low CD42b expression (Figure 1D; P < .002), suggesting loss of the glycocalicin domain. In addition, patient platelets demonstrated decreased levels of sialic acid on platelets compared with similarly prepared control platelets (Figure 1E; P = .003 compared with control). Finally, increased levels of reticulated platelets were found in patients P1 through P5 compared with healthy controls, suggesting increased platelet turnover (Figure 1G). For patient P9, immature platelet fraction was markedly increased compared with healthy controls (55.2% vs reference interval: 1.6% to 4.7%), also consistent with increased platelet turnover. Based on the role of GNE in sialic acid formation and that removal of small amounts of sialic acid residues (8% to 10%) of neuraminidase results in increased platelet clearance,14 we hypothesize that the thrombocytopenia is caused by increased platelet clearance. Although GNE is ubiquitously expressed in all hematologic cells, only platelets are dependent on levels of sialylation. Similarly, in the F2 family, bone marrow aspiration showed increased number of megakaryocytes (Table 1), compatible with increased platelet clearance.15
Immunofluorescence studies of platelets from P5 compared with an unaffected individual’s platelets showed enlarged platelets with normal distribution of surface receptors, cytoskeletal proteins, as well as dense granule, lysosome, and α-granule markers (supplemental Figure 3). The normal distribution of surface receptors by immunofluorescence but decreased expression by flow cytometry is not surprising given that flow cytometry is able to give more information about relative expression levels. The bleeding phenotype in our patients is out of proportion to the degree of thrombocytopenia, suggesting a defect in platelet activation. Therefore, we performed protein analysis of platelet extracts, which demonstrated differential bands in affected individuals by sodium dodecyl sulfate–polyacrylamide gel electrophoresis and mass spectrometry–confirmed downregulation of platelet activation, adhesion, and wound-healing pathways (supplemental Table 1).
There are 2 large registries of GNE myopathy, a Japanese Nationwide patient registry with 121 patients, reporting 3 patients with thrombocytopenia (2.5%), and the Israel Nationwide GNE registry with >200 individuals and no associated thrombocytopenia (Argov Avisohar, Hadassah-Hebrew University Medical Center, e-mail, 2017). In the literature, alterations in platelet count were previously reported in patients with myopathy as an incidental finding, but not as the primary condition.15,16 Isolated thrombocytopenia without associated myopathy due to GNE variants has not been previously reported. Our communication with the UK GAPP investigators identified simultaneously a family of Pakistani origin with homozygous GNE variant.17 We describe 3 additional kindreds with isolated thrombocytopenia and, in the F1 and F2 families reported here, >20 years of follow-up with affected patients with macrothrombocytopenia (up to age 24-42 years) without myopathy. The 1 patient with neurologic findings has a muscle biopsy inconsistent with GNE myopathy (supplemental Figure 1). Thus, we believe that mutations in GNE should be added to the list of diseases associated with isolated macrothrombocytopenia with a bleeding diathesis. It is not clear why some patients have isolated thrombocytopenia, whereas others have myopathy, and further studies are needed to better understand why variants in GNE are associated with 3 distinct clinical phenotypes: myopathy, sialuria,18 and now isolated thrombocytopenia.
In summary, we have defined 3 families with autosomal recessive inheritance of macrothrombocytopenia and mild to life-threatening bleeding histories due to defects in GNE. Notably, these families did not have concurrent muscle wasting phenotypes. The patients appear to have rapid clearance of their platelets associated with loss of their surface GPIb/IX receptors and changes in surface sialylation. Our finding suggests a strong link between sialylation, alteration of surface GPIb/IX expression with reduced GPIX:GPIb expression, increased platelet size, and platelet clearance. Further characterization of the pathways leading to abnormal platelets and/or abnormal muscle biology needs to be defined, and genetic determinants of the observed phenotypic heterogeneity needs to be better understood.
The online version of this article contains a data supplement.
Authorship
Acknowledgments: The authors thank Sara Israels and Eileen McMillan-Ward (technician), Research Institute in Oncology and Hematology, University of Manitoba, Winnipeg, Manitoba, Canada, for their help in the interpretation of the platelet electron microscopic images of F1.
Funding for the NIHR BioResource sequencing project was provided by the National Institute for Health Research (NIHR; grant RG65966). The NIHR BioResource projects were approved by Research Ethics Committees in the United Kingdom and appropriate national ethics authorities in non-UK enrollment centers.
This study makes use of data generated by the NIHR BioResource. A full list of investigators who contributed to the generation of the data is available at https://bioresource.nihr.ac.uk/researchers/researchers/acknowledgement/.
Contribution: S.R.-V., E.S., M.P., D.V., and M.P.L. were involved in study design, interpretation of results, and preparation of the manuscript; E.T. initially identified GNE as the leading candidate gene in F1 and F3; S.R.-V., E.S., D.V., N.J., E.H.-A., G.S., H.D., and Y.K. provided clinical, laboratory, and genetic data of F1; D.J. performed sialylation flow cytometry and interpreted results; K.A. and A.G. provided the immunofluorescence analysis and assisted with interpretation of results; C.K. and S.I. provided clinical, laboratory, and genetic data of F2; M.P.L. provided clinical, laboratory, and genetic data of F3; E.T., K.D., S.V.V.D., R.M., and W.H.O. are members of the NIHR BioResource, who provided clinical and genetic data and played key roles in with interpretation of results. All authors reviewed the manuscript and provided input in manuscript preparation and approved the final draft for submission. The NIHR BioResource–Rare Disease Study is a multicenter whole-genome sequencing study of ∼13 000 patients.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Michele P. Lambert, Children’s Hospital of Philadelphia, Division of Hematology, 3615 Civic Center Blvd, ARC Room 316 G, Philadelphia, PA 19104; e-mail: lambertm@e-mail.chop.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal