

TO THE EDITOR:
Inherited thrombocytopenias are associated with bleeding of all types of severity depending on the reduction in platelet count and whether there is altered platelet function.1 The normal range for platelet counts varies by up to threefold, but an individual’s platelet count is normally maintained within a narrow range. This requires a constant balance between thrombopoiesis and platelet senescence and consumption. Heritable forms of thrombocytopenia are frequently caused by genes that regulate megakaryocytic differentiation and/or platelet production. Next-generation sequencing strategies, such as whole-exome sequencing, are efficient in identifying gene mutations that cause Mendelian disorders.2-4 In this study, we used a whole-exome sequencing approach to elucidate the genetic basis of a severe form of congenital thrombocytopenia.
We present a UK consanguineous family of Pakistani origin with 2 cousins with severe thrombocytopenia (Figure 1B). The proband, III:5, was aged 3 years with a platelet count of 3 × 109/L when entered into the UK Genotyping and Phenotyping of Platelets (GAPP) study. He was born by emergency caesarean section at 34 weeks gestation and had neurological symptoms shortly after birth and bilateral intraventricular hemorrhages. He had a ventriculoperitoneal shunt inserted, which required several revisions, with HLA platelet transfusion prophylaxis. He has developmental delay, skull abnormalities secondary to hydrocephalus, and nystagmus. His baseline platelet count has remained ∼10 × 109/L. He received HLA-matched platelet transfusions every 1 to 2 weeks for the first 12 months of life, and his platelet count increased well thereafter. There were no other abnormalities in the blood count. The bone marrow aspirate and trephine showed a normocellular specimen with normal megakaryocyte numbers and morphology and normal cytogenetics. Patient III:3 was aged 7 years when recruited into the study. She had a baseline platelet count of 15 × 109/L to 20 × 109/L and receives weekly HLA-matched platelet transfusions to minimize symptoms from epistaxis and hematomata, previously causing hospitalization. In both patients, coagulation parameters were normal, and there were no antiplatelet autoantibodies or HLA antibodies. Blood (15 mL) from patients and healthy controls was taken in 10% by volume 3.8% trisodium citrate. Platelet-rich plasma (PRP) was prepared and flow cytometry was conducted as previously described.5 Transmission electron microscopy was performed as described previously6 and examined using a JEOL 1200EX transmission electron microscope. The number of α-granules per square micrometer was calculated for at least 40 platelets from each patient or control. The whole exome of the 2 affected individuals was sequenced with the SureSelect human All Exon 50Mb kit (Agilent Technologies) and sequencing on the HiSeq 2000 (Illumina) with 100-bp paired-end reads. The sequences were aligned to the reference genome (hg19 build).5 To verify candidate mutations, Sanger sequencing was performed using standard methods on an ABI 3730 automated sequencer.
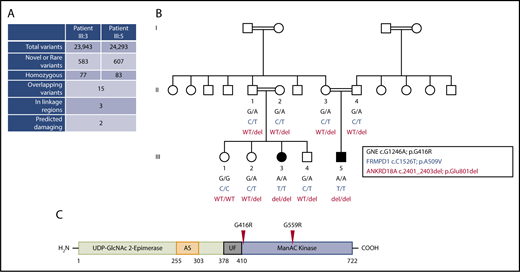
Identification of a homozygous missense substitution in GNE. (A) Filtering strategy of whole-exome sequencing results to identify candidate variants in patients III:3 and III:5 of the same family. (B) Segregation analysis of the exome candidates in family members where DNA was available. The 3 variants (in the genes GNE, FRMPD1, and ANKRD18A) were shared by both affected children and were located within a region of homozygosity on chromosome 9p13.3. Double lines linking parents signify first-cousin unions. (C) Linear domain organization of GNE encoding the enzyme UDP-GlcNAc 2-epimerase/ManNAc kinase. Experimental allosteric sites are based on in vitro studies (AS), region of unknown function (UF). The approximate position of amino acid substitutions (p.G416R and p.G559R) found in the family in this study and in an independent study,11 respectively, are indicated and based on transcript NM_005476. del, deletion; WT, wild-type.
Identification of a homozygous missense substitution in GNE. (A) Filtering strategy of whole-exome sequencing results to identify candidate variants in patients III:3 and III:5 of the same family. (B) Segregation analysis of the exome candidates in family members where DNA was available. The 3 variants (in the genes GNE, FRMPD1, and ANKRD18A) were shared by both affected children and were located within a region of homozygosity on chromosome 9p13.3. Double lines linking parents signify first-cousin unions. (C) Linear domain organization of GNE encoding the enzyme UDP-GlcNAc 2-epimerase/ManNAc kinase. Experimental allosteric sites are based on in vitro studies (AS), region of unknown function (UF). The approximate position of amino acid substitutions (p.G416R and p.G559R) found in the family in this study and in an independent study,11 respectively, are indicated and based on transcript NM_005476. del, deletion; WT, wild-type.
Both patients had severe thrombocytopenia with platelet counts in PRP of 1.5 × 107/mL (patient III:5) and 2.5 × 107/mL (patient III:3), reference range 2.1 × 108/mL to 7.1 × 108/mL (mean ± 2 standard deviation [SD]). Mean platelet volume in patients III:5 and III:3 was 15.0 fL and 10.4 fL, respectively (reference range, 7.68-10.0 fL). Parental platelet counts were normal. An extremely high immature platelet fraction of 87% and 83% (normal range, 1.3% to 10.8%; n = 40) was found in patients III:5 and III:3, respectively, which suggests rapid production (possibly due to rapid clearance). Flow cytometry was used to assess platelet function in the 2 affected individuals using an assay validated for activated platelets with dilutions of PRP from healthy volunteers. The levels of surface glycoproteins CD42b (GPIbα), CD41 (αIIb), and GPVI in patient III:3 were within the reference ranges established in healthy volunteers, whereas for patient III:5, the levels of CD42b and CD41 fell outside these ranges. This could suggest global platelet dysfunction, where loss of glycans can lead to failure of the receptor being transported to the surface or increased proteolysis (Figure 2A). Patient III:5 also showed a complete abolition of CD62P (P-selectin) expression and a very weak increase in binding of fibrinogen to ADP, collagen-related peptide (CRP), and protease-activated receptor 1 (PAR-1) (Figure 2B-C). Patient III:3 showed a slightly greater increase in fibrinogen binding to most agonists than patient III:5 and a recovery of CD62P expression to high concentrations of CRP and PAR-1 peptide, although this was below the range of responses to healthy controls in all cases. Electron microscopy of patient platelets revealed that these are enlarged, but with a similar number of α-granules per surface area compared with controls (Figure 2D).
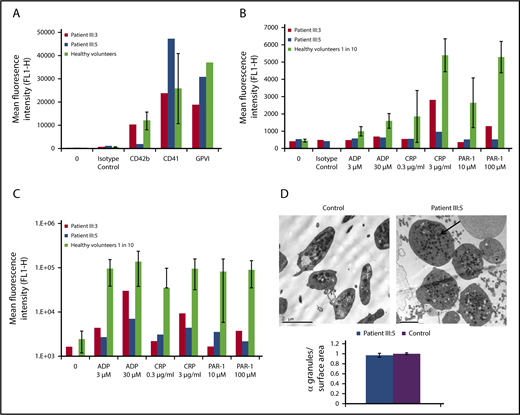
Flow cytometry and transmission electron microscopy assessment of platelet function in patients III:3 and III:5. Flow cytometry assessment of platelet function in patients III:3 and III:5 assessed on an Accuri C6 flow cytometer. (A) Platelet glycoprotein receptors. (B-C) CD62P expression (B) and fluorescent fibrinogen binding (C) following platelet stimulation by various agonists for 2 minutes. The PRP from healthy controls was diluted 1:10 with phosphate-buffered saline and served as a control range. Data for healthy volunteers are shown as mean ± 1 SD (n = 9; except for GPVI, where n = 2). (D) Transmission electron microscopy image of the platelets from patient III:5 and healthy control platelets. Arrow indicates an α-granule, and the graph shows the number of α-granules per surface area. Scale bar, 2 µm. ADP, adenosine 5′-diphosphate.
Flow cytometry and transmission electron microscopy assessment of platelet function in patients III:3 and III:5. Flow cytometry assessment of platelet function in patients III:3 and III:5 assessed on an Accuri C6 flow cytometer. (A) Platelet glycoprotein receptors. (B-C) CD62P expression (B) and fluorescent fibrinogen binding (C) following platelet stimulation by various agonists for 2 minutes. The PRP from healthy controls was diluted 1:10 with phosphate-buffered saline and served as a control range. Data for healthy volunteers are shown as mean ± 1 SD (n = 9; except for GPVI, where n = 2). (D) Transmission electron microscopy image of the platelets from patient III:5 and healthy control platelets. Arrow indicates an α-granule, and the graph shows the number of α-granules per surface area. Scale bar, 2 µm. ADP, adenosine 5′-diphosphate.
The exome of both patients was sequenced, and alignment of the sequencing reads revealed 23 943 and 24 293 variations in patients III:3 and III:5, respectively. Comparisons within the exome variant server, 1000G, and our in-house GAPP database of over 1200 exomes identified 2 homozygous nonsynonymous variants and 1 nonframeshift deletion that were present in both patients (supplemental Tables 1 and 2, available on the Blood Web site), with all variations mapping to a tightly linked homozygous region on chromosome 9p13.3 (supplemental Table 2). The 2 nonsynonymous variants were in genes GNE (p.G416R) and FRMPD1 (p.A509V) and the nonframeshift deletion in ANKRD18A (p.Glu801del). Family studies using Sanger sequencing confirmed that all 3 variants segregated with disease status (Figure 1B). Pathogenicity was predicted using 4 separate in silico–based pathogenicity prediction software programs (MutationTaster, SIFT, PROVEAN, and PolyPhen-2), and conservation at the site of variation was determined using PhyloP and PhastCons. Together, all 3 variants were classified as “unknown significance” when considering the American College of Medical Genetics and Genomics consensus guidelines. Upon further analysis within the ExAC database, only the variants within ANKRD18A and GNE were novel. Data from RNA sequencing of hematopoietic progenitors (blueprint.haem.cam.ac.uk) suggested that there was very low expression of ANKRD18A messenger RNA (mRNA) in hematopoietic progenitors. This is in contrast to GNE mRNA, which is expressed widely in hematopoietic progenitors.
In previous studies, 2 compound heterozygous variations in the gene encoding GNE have been noted to cause a disorder of progressive muscle weakness with a secondary symptom of thrombocytopenia.7,8 Previous dominant mutations in GNE have been associated with sialuria.9,10 It is important to note that recessive patients presented with severe body myopathy as a primary symptom, whereas the patients in our study did not display signs of myopathy, although this is possibly because of their age. Furthermore, a previous study involving whole-exome sequencing of a single pedigree with severe thrombocytopenia and bleeding identified an apparent PRKACG variant, but a strong candidate variant in this family was also a homozygous missense variant in the kinase domain of GNE (p.G559R), as shown in Figure 1C.11
GNE encodes glucosamine (UDP-N-acetyl)-2-epimerase/N-acetylmannosamine kinase, a bifunctional enzyme involved in the sialic acid biosynthesis pathway and is expressed within all cells of the hematopoietic lineage. Thrombocytopenia is known to be associated with increased platelet desialylation in septic patients due to altered platelet production/survival.12 Further, platelet counts were increased in a cohort of influenza patients treated with the sialidase inhibitor oseltamivir (Tamiflu).13 A platelet clearance system has been shown to exist for desialylated platelets containing macrophages and hepatocytes.14 The Ashwell-Morell receptor binds platelets with reduced sialic acid expression,15 and removal of just 8% to 10% of sialic acid residues by neuraminidase treatment leads to increased platelet clearance rates in vivo.16
In summary, our results indicate that the homozygous GNE mutation described here leads to macrothrombocytopenia, possibly due to a reduction in sialic acid biosynthesis, which is expected to cause increased removal of platelets and altered platelet formation.
The online version of this article contains a data supplement.
Acknowledgments
The authors thank the families for providing samples, their clinical and laboratory colleagues for their help, the National Institute for Health Research Haematology Specialty Group for their help in recruiting to the study, and all of their clinical investigators and collaborators.
This work was supported by the British Heart Foundation (PG/13/36/30275, FS/13/70/30521, RG/09/007), a Medical Research Council Doctoral Training Partnership grant (B.J.), a Wellcome Trust Combined Training Programme Fellowship (093994) (G.C.L.), and the Department of Health via the National Institute for Health Research comprehensive Biomedical Research Centre award to Guy’s & St Thomas’ National Health Service Foundation Trust in partnership with King’s College London and King’s College Hospital National Health Service Foundation Trust.
Authorship
Contribution: G.C.L., S.P.W., and N.V.M. designed the research; J.M. and M.W. provided patient samples and clinical data; G.C.L. and N.V.M. undertook the research governance of the study; J.F., A.D., G.C.L., B.J., M.A.S., and N.V.M. performed the research and analyzed data; N.V.M. and A.D. wrote the paper; and all authors critically reviewed and edited the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
A complete list of the members of the UK GAPP Study Group appears in the online appendix.
Correspondence: Neil V. Morgan, Institute of Cardiovascular Sciences, Institute of Biomedical Research, College of Medical and Dental Sciences, University of Birmingham, Edgbaston, Birmingham B15 2TT, United Kingdom; e-mail: n.v.morgan@bham.ac.uk.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal