

Key Points
Hjv−/− mice fail to mount an appropriate hypoferremic response to acute inflammation.
Hjv promotes inflammatory hypoferremia by maintaining a threshold of Bmp6/Smad signaling to hepcidin in hepatocytes.

Abstract
Systemic iron balance is controlled by hepcidin, a liver hormone that limits iron efflux to the bloodstream by promoting degradation of the iron exporter ferroportin in target cells. Iron-dependent hepcidin induction requires hemojuvelin (HJV), a bone morphogenetic protein (BMP) coreceptor that is disrupted in juvenile hemochromatosis, causing dramatic hepcidin deficiency and tissue iron overload. Hjv−/− mice recapitulate phenotypic hallmarks of hemochromatosis but exhibit blunted hepcidin induction following lipopolysaccharide (LPS) administration. We show that Hjv−/− mice fail to mount an appropriate hypoferremic response to acute inflammation caused by LPS, the lipopeptide FSL1, or Escherichia coli infection because residual hepcidin does not suffice to drastically decrease macrophage ferroportin levels. Hfe−/− mice, a model of milder hemochromatosis, exhibit almost wild-type inflammatory hepcidin expression and associated effects, whereas double Hjv−/−Hfe−/− mice phenocopy single Hjv−/− counterparts. In primary murine hepatocytes, Hjv deficiency does not affect interleukin-6 (IL-6)/Stat, and only slightly inhibits BMP2/Smad signaling to hepcidin; however, it severely impairs BMP6/Smad signaling and thereby abolishes synergism with the IL-6/Stat pathway. Inflammatory induction of hepcidin is suppressed in iron-deficient wild-type mice and recovers after the animals are provided overnight access to an iron-rich diet. We conclude that Hjv is required for inflammatory induction of hepcidin and controls the acute hypoferremic response by maintaining a threshold of Bmp6/Smad signaling. Our data highlight Hjv as a potential pharmacological target against anemia of inflammation.
Introduction
Iron entry to the bloodstream is critical for body iron homeostasis and is negatively controlled by hepcidin, the iron-regulatory hormone.1 Hepcidin is secreted from hepatocytes and binds to the iron exporter ferroportin in macrophages, enterocytes, and other iron-releasing target cells. This promotes internalization, ubiquitination, and degradation of ferroportin by lysosomes, which causes iron retention. When systemic iron levels increase, bone morphogenetic protein 6 (BMP6) and to some extent BMP2 trigger transcriptional induction of the hepcidin-encoding HAMP gene via the SMAD pathway; this response prevents iron overload and inhibits dietary iron absorption.2 During infection, interleukin 6 (IL-6) and other inflammatory cytokines promote transcriptional induction of HAMP via STAT signaling; this causes hypoferremia as an innate immune response to deprive invading bacteria from iron, an essential nutrient.3
There is increasing evidence for cross talk between the iron and inflammatory pathways4,5 but the molecular mechanisms are not well understood.6,7 Hemojuvelin (HJV) operates as a BMP coreceptor and is essential for iron-dependent regulation of hepcidin.8 Genetic inactivation of HJV diminishes hepcidin expression and causes severe iron overload (early onset juvenile hemochromatosis) in humans9 and mice.10,11 Nevertheless, Hjv−/− mice appear to retain hepcidin induction following lipopolysaccharide (LPS)-induced inflammation.11,12
HFE, an atypical major histocompatibility complex class I molecule, is another upstream regulator of hepcidin. HFE mutations are associated with the most common form of hereditary hemochromatosis, which presents with milder iron overload and hepcidin deficiency.13 We showed that coablation of Hfe does not affect the iron phenotype of Hjv−/− mice.14 Herein, we use wild-type, Hfe−/−, Hjv−/−, and double Hfe−/−Hjv−/− mice to study hepcidin-associated responses to acute inflammation.
Methods
Animals
Wild-type, Hfe−/−, Hjv−/−, and Hfe−/−Hjv−/− mice were in C57BL/6 background.14 All mice were housed in macrolone cages (up to 5 mice per cage, 12:12 hour light-dark cycle: 7 am to 7 pm; 22°C ± 1°C, 60% ± 5% humidity). Five-week-old male mice were injected intraperitoneally with phosphate-buffered saline, 1 μg/g LPS, or 25 ng/g FSL1. When indicated, mice were fed an iron-deficient diet (2-6 ppm iron)15 or a high-iron diet (2% carbonyl iron).14 Other mice received footpad injection with 108 colony-forming units (CFU) of Escherichia coli SP15.6 At the end points, all animals were sacrificed by CO2 inhalation. Experimental procedures were approved by the animal care committee of McGill University (protocol 4966).
Hepatocyte culture
Primary hepatocytes were isolated from livers of wild-type and Hjv−/− mice (22-25 g) by using a 2-step collagenase perfusion technique16 with some modifications (see supplemental Methods, available on the Blood Web site). The cells were seeded onto tissue-culture dishes (26 000 cells/cm2) in Williams’ medium E (Gibco) supplemented with 10% heat-inactivated fetal bovine serum, 2 mM l-glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin, and allowed 90 minutes to attach. The serum-containing medium was then removed, and the cells were subjected to different culture conditions in serum-free Williams’ medium E. In control groups, cells were incubated with medium alone for the indicated time of the experiment.
Materials
LPS (serotype 055:B5) was purchased from Sigma-Aldrich and FSL1 (Pam2CGDPKHPKSF) from InvivoGen. The following recombinant proteins were used: murine IL-6 (Cell Signaling), human IL-6 (Sigma-Aldrich), human/mouse/rat BMP2 (R&D Systems), human BMP6 (R&D Systems), and human holotransferrin (Sigma-Aldrich).
Serum biochemistry
Blood was collected via cardiac puncture. Serum was prepared by using micro Z-gel tubes with clotting activator (Sarstedt) and was snap-frozen at −80°C. Serum iron and total iron-binding capacity (TIBC) were determined using a Roche Hitachi 917 Chemistry Analyzer. Transferrin saturation was calculated from the ratio of serum iron and TIBC. Serum hepcidin was measured by using an ELISA kit (Intrinsic LifeSciences).
Liver iron content
Liver iron was quantified by the ferrozine assay.17
Biochemical assays and histology
Quantitative real-time polymerase chain reaction (qPCR), western blotting, and immunohistochemistry were performed as described.17-19 Details are provided in the supplemental Methods.
Statistical analysis
Statistical analysis was performed by using the Prism GraphPad software (version 7.0c). Multiple groups were subjected to analysis of variance (ANOVA) with Bonferroni posttest comparison. A probability value P < .05 was considered statistically significant.
Results
Kinetics of LPS-induced inflammatory responses in wild-type mice
Wild-type mice were used to characterize pathophysiological responses associated with acute inflammatory induction of hepcidin. The animals were left untreated or injected with LPS and euthanized after 2, 4, 8, 18, or 24 hours. Another group of mice was fed for 5 days with a high-iron diet to obtain reference values for iron-mediated hepcidin responses. As anticipated,20 the LPS treatment triggered rapid induction of Il6 messenger RNA (mRNA) (Figure 1A), as well as robust Stat3 phosphorylation (Figure 1B) in the liver. These responses peaked at 2 hours and gradually attenuated afterward, with Il6 mRNA returning to baseline after 18 hours. Activation of the Il6/Stat-signaling pathway by LPS correlated with increased Hamp mRNA expression that peaked (3.8-fold; P < .0001) within 4 hours and normalized after 8 hours (Figure 1D). This caused a 2.9-fold (P < .0001) increase of serum hepcidin at 4 hours (Figure 1E). The peak Hamp mRNA and serum hepcidin values in LPS-treated animals approach the respective levels obtained following dietary iron loading (indicated by dotted lines).
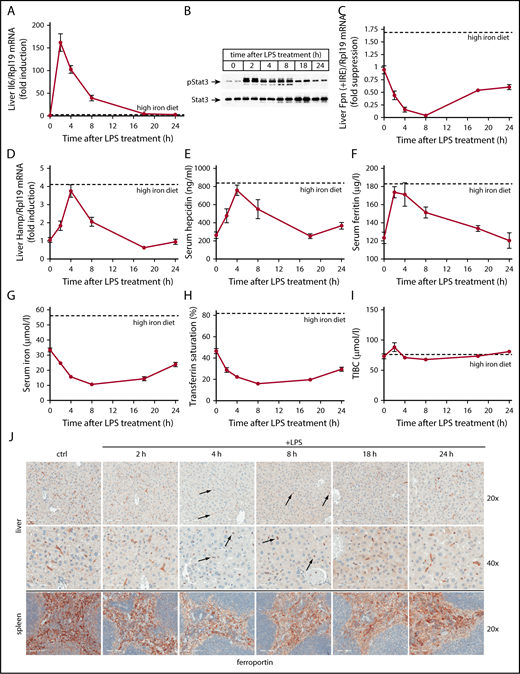
Kinetic analysis of LPS-induced inflammatory responses in wild-type mice. Five-week-old male wild-type C57BL/6 mice (n = 6 for each experimental group) were left untreated or injected with 1 μg/g LPS and sacrificed after 2, 4, 8, 18, or 24 hours. Sera were used for iron biochemistry, and tissues were processed for preparation of RNA and protein extracts or fixed for histology. (A) qPCR analysis of liver Il6 mRNA; (B) western blot analysis of liver pStat3 and Stat3; (C) qPCR analysis of liver ferroportin (Fpn + IRE) mRNA (IRE-containing isoform); (D) qPCR analysis of liver Hamp mRNA; (E) serum hepcidin; (F) serum ferritin; (G) serum iron; (H) transferrin saturation; (I) TIBC. All data in the graphs (A-I) are presented as the mean plus or minus standard error of the mean (SEM). Dotted lines indicate average values obtained from age-matched male C57BL/6 mice (n = 3) fed a high-iron diet for 5 days. (J) Immunohistochemical staining of ferroportin in liver and spleen sections (original magnification ×20 and ×40). Arrows indicate ferroportin internalization in Kupffer cells.
Kinetic analysis of LPS-induced inflammatory responses in wild-type mice. Five-week-old male wild-type C57BL/6 mice (n = 6 for each experimental group) were left untreated or injected with 1 μg/g LPS and sacrificed after 2, 4, 8, 18, or 24 hours. Sera were used for iron biochemistry, and tissues were processed for preparation of RNA and protein extracts or fixed for histology. (A) qPCR analysis of liver Il6 mRNA; (B) western blot analysis of liver pStat3 and Stat3; (C) qPCR analysis of liver ferroportin (Fpn + IRE) mRNA (IRE-containing isoform); (D) qPCR analysis of liver Hamp mRNA; (E) serum hepcidin; (F) serum ferritin; (G) serum iron; (H) transferrin saturation; (I) TIBC. All data in the graphs (A-I) are presented as the mean plus or minus standard error of the mean (SEM). Dotted lines indicate average values obtained from age-matched male C57BL/6 mice (n = 3) fed a high-iron diet for 5 days. (J) Immunohistochemical staining of ferroportin in liver and spleen sections (original magnification ×20 and ×40). Arrows indicate ferroportin internalization in Kupffer cells.
The transient LPS-induced hepcidinemia promoted a more sustained hypoferremia, with a maximal 3.3-fold (P < .0001) and 2.9-fold (P < .0001) drop of serum iron (Figure 1G) and transferrin saturation (Figure 1H), respectively, 8 hours post-LPS administration. Serum iron and transferrin saturation remained low even after 18 hours, in spite of serum hepcidin normalization. TIBC was slightly augmented by LPS at 2 hours, but immediately returned to baseline and remained largely unaffected afterward (Figure 1I). Serum ferritin, a marker of iron and inflammation,21 was raised by 40% (P < .01) 2 hours after LPS treatment, and normalized within 24 hours (Figure 1F).
Ferroportin mRNA dramatically declined in livers of LPS-treated mice (Figure 1C), as expected.22 A nadir of 95% suppression (P < .0001) was reached after 8 hours. Immunohistochemical analysis confirms previous findings10,23 that liver ferroportin is primarily expressed in c-fms+ Kupffer cells (supplemental Figure 1). LPS promoted a considerable time-dependent change in the expression pattern. Thus, 4 to 8 hours following LPS treatment, ferroportin staining shifted from elongated (due to dendritic branching) plasma membranes of Kupffer cells to round intracellular compartments (Figure 1J top, see arrows). Plasma membrane expression of ferroportin progressively recovered afterward. Quantification with ImageScope software revealed a maximal ∼80% decrease in ferroportin signal intensity in response to LPS.
Splenic ferroportin is primarily expressed in c-fms+ red pulp macrophages10,23 (supplemental Figure 1). LPS decreased signal intensity by ∼30% within 4 to 8 hours (Figure 1J bottom). The downregulation of ferroportin in the liver and spleen of LPS-treated mice (Figure 1J) concurs with the hepcidinemia peak (Figure 1E), suggesting that it represents hepcidin-dependent ferroportin internalization and degradation.
Hjv deficiency impairs hepcidin-dependent responses to LPS-induced acute inflammation
To evaluate the roles of Hfe and Hjv in inflammatory regulation of hepcidin, wild-type, Hfe−/−, Hjv−/−, and Hfe−/−Hjv−/− mice were injected with LPS for 4 hours. As expected,14,24 basal serum iron levels and transferrin saturation were elevated in Hfe−/− and further increased in Hjv−/− and Hfe−/−Hjv−/− saline-injected control mice (Figure 2A-B), whereas TIBC was unaltered (Figure 2C). LPS triggered a statistically significant decline in serum iron and transferrin saturation in all genotypes. This effect was dramatic in wild-type and Hfe−/− mice (>50%), and only modest (∼15%) in Hjv−/− and Hfe−/−Hjv−/− mice. Thus, the hypoferremic response to acute inflammation is not compromised in the absence of Hfe but is severely blunted by Hjv deficiency.
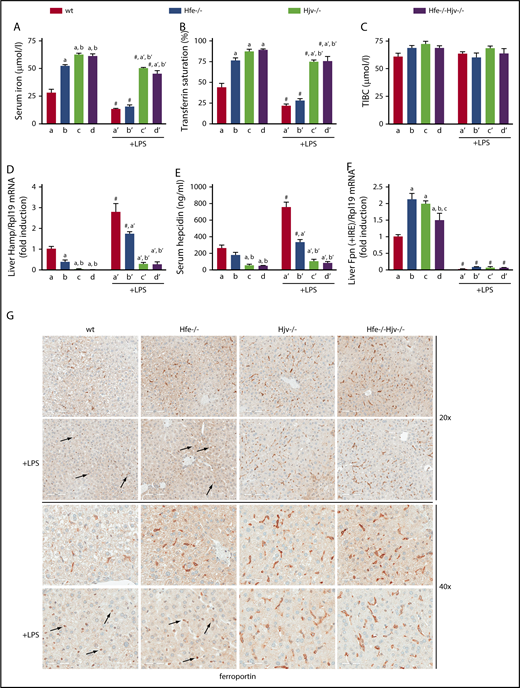
Hjv deficiency abrogates hepcidin-dependent ferroportin internalization and hypoferremic response to acute inflammation. Five-week-old male wild-type (wt), Hfe−/−, Hjv−/−, and double Hfe−/−Hjv−/− mice (all in C57BL/6 background; n = 6 for each experimental group) were injected with either phosphate-buffered saline or 1 μg/g LPS. After 4 hours, all animals were sacrificed; sera were used for iron biochemistry, and tissues were processed for preparation of RNA and protein extracts or fixed for histology. (A) Serum iron; (B) transferrin saturation; (C) TIBC; (D) qPCR analysis of liver Hamp mRNA; (E) serum hepcidin; (F) qPCR analysis of liver ferroportin (Fpn + IRE) mRNA (IRE-containing isoform). All data are presented as the mean plus or minus SEM. Statistical analysis was performed by 2-way ANOVA. Statistically significant differences (P < .05) across genotypes (vs columns a, b, c, a′, b′) are indicated by a, b, c, a′, b′, and across LPS treatment by the pound sign (#). (G) Immunohistochemical staining of ferroportin in liver sections (original magnification ×20 and ×40). Arrows indicate ferroportin internalization in Kupffer cells from LPS-treated wt and Hfe−/− mice.
Hjv deficiency abrogates hepcidin-dependent ferroportin internalization and hypoferremic response to acute inflammation. Five-week-old male wild-type (wt), Hfe−/−, Hjv−/−, and double Hfe−/−Hjv−/− mice (all in C57BL/6 background; n = 6 for each experimental group) were injected with either phosphate-buffered saline or 1 μg/g LPS. After 4 hours, all animals were sacrificed; sera were used for iron biochemistry, and tissues were processed for preparation of RNA and protein extracts or fixed for histology. (A) Serum iron; (B) transferrin saturation; (C) TIBC; (D) qPCR analysis of liver Hamp mRNA; (E) serum hepcidin; (F) qPCR analysis of liver ferroportin (Fpn + IRE) mRNA (IRE-containing isoform). All data are presented as the mean plus or minus SEM. Statistical analysis was performed by 2-way ANOVA. Statistically significant differences (P < .05) across genotypes (vs columns a, b, c, a′, b′) are indicated by a, b, c, a′, b′, and across LPS treatment by the pound sign (#). (G) Immunohistochemical staining of ferroportin in liver sections (original magnification ×20 and ×40). Arrows indicate ferroportin internalization in Kupffer cells from LPS-treated wt and Hfe−/− mice.
Liver Hamp mRNA inversely correlated with serum iron and transferrin saturation in all genotypes (Figure 2D), as reported.14 LPS promoted approximately threefold Hamp mRNA induction in wild-type and Hfe−/− mice. LPS-mediated upregulation of Hamp mRNA was even stronger (more than sixfold) in Hjv−/− and Hfe−/−Hjv−/− mice. However, because basal Hamp expression was negligible, LPS-treated Hjv−/− and Hfe−/−Hjv−/− mice expressed lower Hamp mRNA than untreated wild-type and Hfe−/− counterparts. An almost identical picture emerges from the quantification of serum hepcidin in all genotypes, before and after LPS treatment (Figure 2E).
LPS profoundly suppressed hepatic ferroportin (Fpn plus iron-responsive element [IRE]) mRNA in all genotypes (Figure 2F). The LPS treatment caused internalization of ferroportin in Kupffer cells from wild-type and Hfe−/−, but only marginally from Hjv−/− and Hfe−/−Hjv−/−, mice (Figure 2G). Image quantification revealed ∼80% loss of ferroportin signal intensity in wild-type and Hfe−/− livers, and ∼10% loss in Hjv−/− and Hfe−/−Hjv−/− livers. Strong ferroportin expression (∼90% of control) was also preserved in splenic macrophages of LPS-treated Hjv−/− and Hfe−/−Hjv−/− mice (supplemental Figure 2). Image quantification showed ∼30% loss of ferroportin signal intensity in wild-type and Hfe−/− spleens, and ∼10% loss in Hjv−/− and Hfe−/−Hjv−/− spleens. LPS-mediated hepatic induction of the inflammatory cytokines Il6, Tnfa, and Il1b was similar in all genotypes (supplemental Figure 3), indicating that the lack of Hjv (or Hfe) does not interfere with the respective pathways.
A kinetic analysis over 24 hours further validates the defective hypoferremic response of Hjv−/− and Hfe−/−Hjv−/− mice to LPS (supplemental Figure 4A-C), which is largely attributable to blunted Hamp mRNA induction (supplemental Figure 4D). These data also show that LPS not only suppresses liver ferroportin mRNA (supplemental Figure 4E), but also duodenal ferroportin (supplemental Figure 4F) and Dmt1 (supplemental Figure 4G) mRNAs in all genotypes, in agreement with published data.22,25
Hjv deficiency impairs inflammatory hypoferremic response to E coli infection or FSL1 treatment
To examine the biological significance of Hjv as an upstream inflammatory regulator of hepcidin, Hjv−/− and wild-type mice were infected with E coli via footpad injection. This resulted in a rapid drop in serum iron and transferrin saturation within 4 hours, which persisted over 24 hours (Figure 3A-C). Hamp mRNA and serum hepcidin were induced in both genotypes, but levels were considerably higher in wild-type mice (Figure 3D-E). Interestingly, hepcidin induction in E coli–infected wild-type mice appeared less pronounced compared with LPS-treated counterparts (compare Figures 1D-E and 2D-E with Figure 3D-E). Moreover, serum hepcidin values in E coli–infected Hjv−/− mice approached those of untreated control mice (Figure 3E). Nevertheless, serum iron remained supraphysiologically elevated in the Hjv−/− mice (Figure 3A), very likely because these animals express higher basal ferroportin levels than controls (Figure 2G). Liver ferroportin mRNA was equally suppressed in both genotypes (Figure 3F). The data with this infection model highlight the critical role of Hjv in the hepcidin-mediated hypoferremic response to acute inflammation.
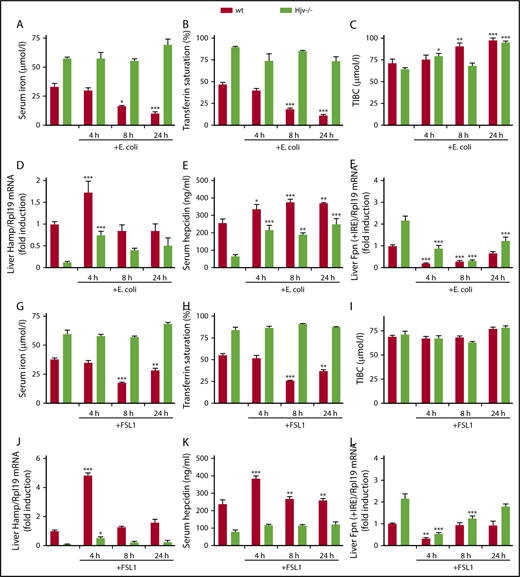
Hjv−/−mice exhibit defective hypoferremic response to infection with E coli or treatment with FSL1. Five-week-old male wt and Hjv−/− mice (n = 6 for each experimental group) were either left untreated or received a footpad injection with 108 CFU E coli SP15, or an intraperitoneal injection with 25 ng/g FSL1, and euthanized after 4, 8, or 24 hours. Sera were used for iron biochemistry and livers were processed for preparation of RNA. (A,G) Serum iron; (B,H) transferrin saturation; (C,I) TIBC; (D,J) qPCR analysis of liver Hamp mRNA; (E,K) serum hepcidin; and (F,L) qPCR analysis of liver ferroportin (Fpn + IRE) mRNA in response to E coli infection (A-F) or FSL1 treatment (G-L). All data are presented as the mean plus or minus SEM. Statistical analysis was performed by 2-way ANOVA. Statistically significant differences between E coli–infected or FSL1-treated mice of each genotype and respective untreated controls are indicated by * (P < .05), ** (P < .01), or *** (P < .001).
Hjv−/−mice exhibit defective hypoferremic response to infection with E coli or treatment with FSL1. Five-week-old male wt and Hjv−/− mice (n = 6 for each experimental group) were either left untreated or received a footpad injection with 108 CFU E coli SP15, or an intraperitoneal injection with 25 ng/g FSL1, and euthanized after 4, 8, or 24 hours. Sera were used for iron biochemistry and livers were processed for preparation of RNA. (A,G) Serum iron; (B,H) transferrin saturation; (C,I) TIBC; (D,J) qPCR analysis of liver Hamp mRNA; (E,K) serum hepcidin; and (F,L) qPCR analysis of liver ferroportin (Fpn + IRE) mRNA in response to E coli infection (A-F) or FSL1 treatment (G-L). All data are presented as the mean plus or minus SEM. Statistical analysis was performed by 2-way ANOVA. Statistically significant differences between E coli–infected or FSL1-treated mice of each genotype and respective untreated controls are indicated by * (P < .05), ** (P < .01), or *** (P < .001).
We investigated whether inflammatory suppression of ferroportin mRNA may trigger hypoferremia in a hepcidin-independent manner. To this end, Hjv−/− and wild-type mice were injected with the lipopeptide FSL1, a Toll-like receptor 2/6 (TLR2/6) ligand reported to cause hypoferremia by negatively regulating ferroportin mRNA.26 We expected that FSL1 would decrease serum iron in Hjv−/− mice, similar to wild-type controls. However, Hjv−/− mice were resistant to FSL1-induced hypoferremia (Figure 3G-I), even though liver ferroportin mRNA was appropriately suppressed (Figure 3L). Surprisingly, the hypoferremic response in wild-type mice, which peaked at 8 hours (Figure 3G), was accompanied by significant hepcidin induction (Figure 3J-K). Conceivably, this was driven by IL-6 (supplemental Figure 5), a target of TLR2/6 signaling in macrophages,27 but more conclusive evidence would require experiments with Il6−/− mice. These data appear to exclude a major contribution of ferroportin mRNA suppression to acute inflammation in Hjv−/− mice, at least under our experimental conditions.
Hjv promotes inflammatory induction of hepcidin by maintaining active Bmp6-dependent Smad signaling
Using the LPS model, we explored mechanisms by which Hjv modulates inflammatory signaling to hepcidin. Stat3 phosphorylation was undetectable in livers of saline-treated mice and was profoundly stimulated by LPS in all genotypes (Figure 4A). This was also associated with high expression of Socs3 mRNA (Figure 4B), a Stat3 target. Basal Smad5 phosphorylation was robust in wild-type and Hfe−/− livers (with some variability) but diminished in Hjv−/− and Hfe−/−Hjv−/− livers (Figure 4A), consistent with Hamp expression. Following LPS treatment, hepatic Smad5 phosphorylation was induced in all genotypes, and this correlated with upregulation of Inhbb mRNA (Figure 4C). LPS decreased hepatic Bmp6 mRNA (Figure 4D) and did not appear to significantly affect expression of the Bmp/Smad target gene Id1 in all genotypes (Figure 4E), in line with previous observations in wild-type mice.6
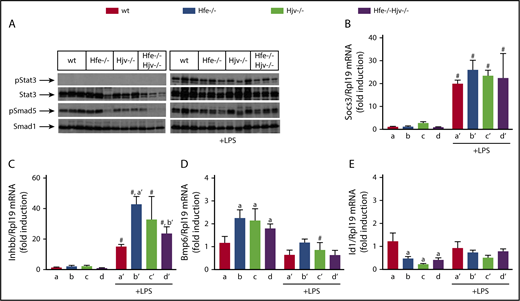
Livers of Hjv-deficient mice retain inflammatory Smad signaling, presumably via activin B. (A-E) Liver protein extracts and RNA from mice described in Figure 2 were used for western blotting and qPCR analysis, respectively. (A) Levels of pStat3, Stat3, pSmad5, and Smad5; (B) Socs3 mRNA; (C) Inhbb mRNA; (D) Bmp6 mRNA; (E) Id1 mRNA. All data are presented as the mean plus or minus SEM. Statistical analysis was performed by 2-way ANOVA. Statistically significant differences (P < .05) across genotypes (vs columns a, a′, b′) are indicated by a, a′, b′, and across LPS treatment by pound sign (#).
Livers of Hjv-deficient mice retain inflammatory Smad signaling, presumably via activin B. (A-E) Liver protein extracts and RNA from mice described in Figure 2 were used for western blotting and qPCR analysis, respectively. (A) Levels of pStat3, Stat3, pSmad5, and Smad5; (B) Socs3 mRNA; (C) Inhbb mRNA; (D) Bmp6 mRNA; (E) Id1 mRNA. All data are presented as the mean plus or minus SEM. Statistical analysis was performed by 2-way ANOVA. Statistically significant differences (P < .05) across genotypes (vs columns a, a′, b′) are indicated by a, a′, b′, and across LPS treatment by pound sign (#).
Bmp628 and Bmp229 are physiological ligands for signaling to Hamp, and Bmp6 appears to be essential for both iron30 and inflammatory12 pathways. Recombinant BMP6 and IL-6 were reported to synergistically induce HAMP in HepG2 cells.5 We validated this in Huh7 hepatoma cells (supplemental Figure 6). Moreover, supplemental Figure 6 shows that BMP2 likewise synergizes with IL-6 for HAMP induction, whereas addition of BMP2 to BMP6 or BMP6/IL-6 does not further stimulate HAMP.
Next, we investigated the roles of Hjv, BMP6, and BMP2 in experiments with primary wild-type and Hjv−/− hepatocytes. The cells were cultured in serum-free media, where basal hepcidin expression is suppressed.31 Treatment with BMP6 for 4 hours strongly induced Hamp mRNA and Smad5 phosphorylation in wild-type but not Hjv−/− hepatocytes (Figure 5A-B). These responses were not affected by iron-loaded transferrin (holo-transferrin [holo-Tf]), which in its own right modestly stimulated Hamp mRNA in wild-type but not Hjv−/− hepatocytes, as reported.32 IL-6 alone triggered a likewise modest activation of Hamp mRNA, but in both wild-type and Hjv−/− hepatocytes (Figure 5A), which was associated with Stat3 phosphorylation (Figure 5B) and induction of downstream Socs3 mRNA (Figure 5D).
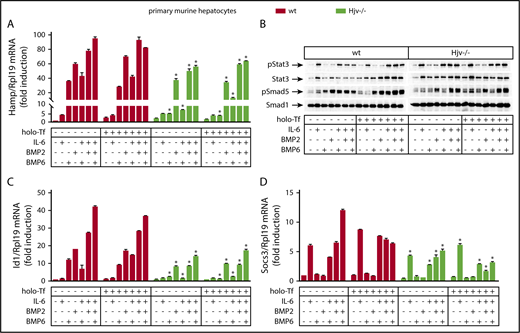
Hjv−/− hepatocytes exhibit impaired Smad signaling in response to BMP6. Primary hepatocytes were isolated from livers of wt and Hjv−/− mice. The cells were cultured in serum-free media and left untreated or pretreated overnight with 30 μM holo-Tf. On the next day, the cells were supplemented (or not) without previous wash with 20 ng/mL murine IL-6, 5 ng/mL human BMP6, or 5 ng/mL human/mouse/rat BMP2, alone or in combinations, as indicated. The incubation was terminated after 4 hours and the cells were harvested and lysed. Hepatocyte RNA and protein extracts were analyzed by qPCR and western blotting, respectively. (A) Expression of Hamp mRNA; (B) levels of pStat3, Stat3, pSmad5, and Smad5; (C) Id1 mRNA; (D) Socs3 mRNA. All data are presented as the mean plus or minus SEM. Statistical analysis was performed by 2-way ANOVA. Statistically significant differences (P < .05) across genotypes are indicated by an asterisk (*).
Hjv−/− hepatocytes exhibit impaired Smad signaling in response to BMP6. Primary hepatocytes were isolated from livers of wt and Hjv−/− mice. The cells were cultured in serum-free media and left untreated or pretreated overnight with 30 μM holo-Tf. On the next day, the cells were supplemented (or not) without previous wash with 20 ng/mL murine IL-6, 5 ng/mL human BMP6, or 5 ng/mL human/mouse/rat BMP2, alone or in combinations, as indicated. The incubation was terminated after 4 hours and the cells were harvested and lysed. Hepatocyte RNA and protein extracts were analyzed by qPCR and western blotting, respectively. (A) Expression of Hamp mRNA; (B) levels of pStat3, Stat3, pSmad5, and Smad5; (C) Id1 mRNA; (D) Socs3 mRNA. All data are presented as the mean plus or minus SEM. Statistical analysis was performed by 2-way ANOVA. Statistically significant differences (P < .05) across genotypes are indicated by an asterisk (*).
Contrary to BMP6, BMP2 induced Hamp mRNA and Smad5 phosphorylation in both wild-type and Hjv−/− hepatocytes, yet with ∼35% lower efficiency in the latter (Figure 5A-B). This is also reflected in levels of Id1 mRNA, a target of Bmp/Smad signaling (Figure 5C). The responses to BMP2 were not affected by holo-Tf.
Combinations of IL-6 with either BMP6 or BMP2 resulted in somehow less pronounced synergism for Hamp mRNA induction in wild-type primary hepatocytes compared with Huh7 hepatoma cells and did not appear to further affect Smad5 or Stat3 phosphorylation. As in hepatoma cells, combined administration of both BMP2 and BMP6 did not additionally stimulate Hamp in the presence or absence of IL-6. These data suggest that Hjv enhances inflammatory induction of Hamp mRNA by maintaining active Smad signaling via primarily Bmp6.
In a prolonged (18-hour) incubation, higher doses of BMP6 were able to at least partially induce Hamp mRNA and Smad5 phosphorylation in Hjv−/− hepatocytes, yet this effect was significantly weaker compared with that achieved by equivalent doses of BMP2 (supplemental Figure 7). Under these conditions, Hamp mRNA levels were highly induced in wild-type, but remained stationary in Hjv−/− hepatocytes exposed to either BMP6 or BMP2 or both, with or without IL-6.
Hepcidin-induced hypoferremic response to LPS requires adequate hepatic iron
To investigate whether hepatic iron content affects inflammatory induction of hepcidin and the ensuing hypoferremic response, wild-type mice were placed on an iron-deficient diet for 3 weeks. Subsequently, half of the animals were switched overnight (12 hours) to a high-iron diet and on the next day, all mice were treated with LPS (or saline) for 4 hours. The iron-deficient diet caused hypoferremia; this was not merely corrected by overnight exposure to the high-iron diet but progressed to hyperferremia (Figure 6A-C). Likewise, iron-deficient mice had reduced hepatic iron content, which was normalized by the overnight iron supplementation (Figure 6D). The LPS challenge did not further reduce serum iron levels in iron-deficient mice but promoted a robust hypoferremic response in iron-supplemented animals. Consistently, Hamp mRNA expression was undetectable in iron-deficient mice and remained marginal even after LPS stimulation (Figure 6E), in agreement with earlier findings.33 Likewise, serum hepcidin remained below physiological levels under these conditions (Figure 6F). The evidently residual induction of hepcidin failed to cause ferroportin internalization in Kupffer cells (Figure 6G). On the other hand, dietary iron supplementation restored basal Hamp mRNA and serum hepcidin. This allowed further hepcidin induction by LPS, which promoted ferroportin internalization in Kupffer cells (Figure 6G right) and splenic macrophages (supplemental Figure 8). Thus, inflammatory induction of hepcidin is effective and elicits downstream biologic responses in mice with adequate hepatic iron content.
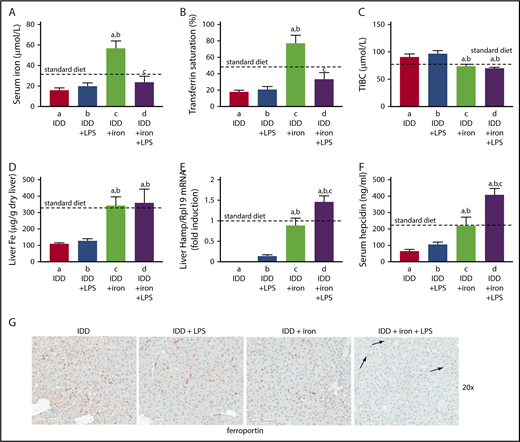
Hepcidin-induced ferroportin internalization and hypoferremic response to acute inflammation requires adequate liver iron. Eight-week-old male C57BL/6 mice (n = 24) were fed with an iron-deficient diet for 3 weeks. During the last night, half of the animals were provided a high-iron diet (2% carbonyl iron). On the next day, all mice (n = 6 for each group) were injected with either phosphate-buffered saline or 1 μg/g LPS and sacrificed after 4 hours. (A) Serum iron; (B) transferrin saturation; (C) TIBC; (D) liver iron content; (E) liver Hamp mRNA; (F) serum hepcidin. All data are presented as the mean plus or minus SEM. Dotted lines indicate average values obtained from age-matched male C57BL/6 mice (n = 3) on standard diet. Statistical analysis was performed by 1-way ANOVA. Statistically significant differences (P < .05) across treatments (vs columns a, b, c) are indicated by a, b, c. (G) Immunohistochemical staining of ferroportin in liver sections (original magnification ×20). Arrows indicate ferroportin internalization in Kupffer cells from LPS-treated mice that had overnight access to the high-iron diet.
Hepcidin-induced ferroportin internalization and hypoferremic response to acute inflammation requires adequate liver iron. Eight-week-old male C57BL/6 mice (n = 24) were fed with an iron-deficient diet for 3 weeks. During the last night, half of the animals were provided a high-iron diet (2% carbonyl iron). On the next day, all mice (n = 6 for each group) were injected with either phosphate-buffered saline or 1 μg/g LPS and sacrificed after 4 hours. (A) Serum iron; (B) transferrin saturation; (C) TIBC; (D) liver iron content; (E) liver Hamp mRNA; (F) serum hepcidin. All data are presented as the mean plus or minus SEM. Dotted lines indicate average values obtained from age-matched male C57BL/6 mice (n = 3) on standard diet. Statistical analysis was performed by 1-way ANOVA. Statistically significant differences (P < .05) across treatments (vs columns a, b, c) are indicated by a, b, c. (G) Immunohistochemical staining of ferroportin in liver sections (original magnification ×20). Arrows indicate ferroportin internalization in Kupffer cells from LPS-treated mice that had overnight access to the high-iron diet.
Although expression of the inflammatory cytokines Il6, Tnfa, and Il1b was not affected by iron (supplemental Figure 9), iron-deficient livers exhibited reduced Smad5 phosphorylation (Figure 7A) and Bmp6 mRNA expression (Figure 7D). Interestingly, phospho-Smad5 levels remained low even after LPS stimulation, in spite of Inhbb mRNA induction (Figure 7C). Iron supplementation boosted Smad5 phosphorylation and Bmp6 mRNA, and appropriately modulated expression of the iron markers transferrin receptor 1 (TfR1) and ferritin. Under these conditions, LPS induced Inhbb mRNA, suppressed Bmp6 mRNA, and did not affect phospho-Smad5 levels. LPS promoted similar Stat3 phosphorylation and Socs3 mRNA induction in iron-deficient and -supplemented mice (Figure 7B), regardless of hepatic iron content. In accord with recent data,6 the Smad targets Id1 and Smad7 mRNAs were only induced by iron but not LPS. Moreover, LPS inhibited expression of Smad7 in iron-replete livers. LPS also severely suppressed ferroportin mRNAs (+IRE and -IRE isoforms; Figure 7G-H). The effective inflammatory induction of hepcidin in iron-replete livers (Figure 6E-F) under conditions of Bmp6 mRNA suppression (Figure 7D) raises the possibility for direct regulation of the pathway by further signals, possibly related to the hepatic iron content.
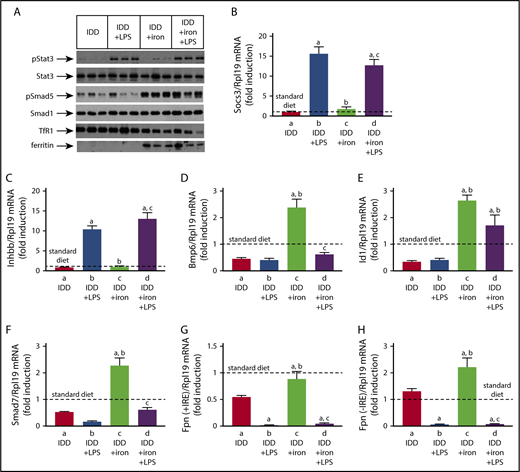
Smad and Stat signaling in LPS-treated mice with variable liver iron content. Liver protein extracts and RNA from mice described in Figure 6 were used for western blotting and qPCR analysis, respectively. (A) Levels of pStat3, Stat3, pSmad5, Smad5, TfR1, and ferritin; (B) Socs3 mRNA; (C) Inhbb mRNA; (D) Bmp6 mRNA; (E) Id1 mRNA; (F) Smad7 mRNA; (G) (Fpn + IRE) mRNA (IRE-containing isoform); (H) (Fpn − IRE) mRNA (isoform lacking IRE). All data are presented as the mean plus or minus SEM. Dotted lines indicate average values obtained from age-matched male C57BL/6 mice (n = 3) on standard diet. Statistical analysis was performed by 1-way ANOVA. Statistically significant differences (P < .05) across treatments (vs columns a, b, c) are indicated by a, b, c.
Smad and Stat signaling in LPS-treated mice with variable liver iron content. Liver protein extracts and RNA from mice described in Figure 6 were used for western blotting and qPCR analysis, respectively. (A) Levels of pStat3, Stat3, pSmad5, Smad5, TfR1, and ferritin; (B) Socs3 mRNA; (C) Inhbb mRNA; (D) Bmp6 mRNA; (E) Id1 mRNA; (F) Smad7 mRNA; (G) (Fpn + IRE) mRNA (IRE-containing isoform); (H) (Fpn − IRE) mRNA (isoform lacking IRE). All data are presented as the mean plus or minus SEM. Dotted lines indicate average values obtained from age-matched male C57BL/6 mice (n = 3) on standard diet. Statistical analysis was performed by 1-way ANOVA. Statistically significant differences (P < .05) across treatments (vs columns a, b, c) are indicated by a, b, c.
Discussion
We evaluated the role of Hjv on inflammatory induction of hepcidin and the hypoferremic response to acute inflammation. In an exploratory time-course experiment, we show that LPS triggers rapid upregulation of liver Hamp mRNA in wild-type mice, which peaks at 4 hours (Figure 1D) and is apparently linked to the earlier activation of Il6/Stat signaling (Figure 1A-B). This response is accompanied by an increase in serum hepcidin (Figure 1E) with kinetics accurately mirroring Hamp mRNA. A good agreement between serum hepcidin and Hamp mRNA has also been observed in other settings.12,24,34
LPS-induced hepcidinemia temporally correlates with decreased ferroportin expression in Kupffer cells and splenic macrophages (Figure 1J), which recycle iron from senescent red blood cells.1 Ferroportin downregulation is apparently due to hepcidin-dependent degradation and is consistent with its visible internalization in Kupffer cells. These responses are associated with hypoferremia (Figure 1G-H) that persists even after normalization of serum hepcidin levels, under conditions where macrophage ferroportin expression gradually recovers. Our results underline the significance of the hepcidin/ferroportin axis in inflammatory hypoferremia and are largely consistent with earlier kinetic data in LPS-treated mice34,35 or humans.36
We examined LPS-induced hypoferremic responses in models of hemochromatosis. Hfe−/− mice manifested efficient induction of Hamp mRNA and developed hepcidinemia, which promoted hypoferremia via ferroportin degradation in Kupffer cells (Figure 2). Even though Hamp mRNA upregulation was weaker and serum hepcidin lower compared with wild-type mice, the degradation of macrophage ferroportin and the drop of serum iron were indistinguishable. These data corroborate that Hfe is dispensable for Hamp induction by the inflammatory pathway,37-39 contrary to an earlier opposite conclusion.40 Moreover, they suggest that the development of hypoferremia requires a minimal effective concentration of serum hepcidin.
Apparently, the hepcidin threshold is not reached in Hjv−/− and Hfe−/−Hjv/− mice. These animals mount a considerable more than sixfold inflammatory induction of Hamp. However, basal Hamp mRNA levels are minuscule and remain low even following induction, failing to promote substantial hepcidin accumulation. Thus, the residual inflammatory induction of hepcidin in Hjv-deficient mice cannot elicit extensive ferroportin degradation and iron retention in macrophages, and therefore appears to have limited biological significance. Essentially, Hjv−/− and Hfe−/−Hjv−/− mice remain hyperferremic following LPS treatment. The small LPS-mediated drop in serum iron in these animals may result from the residual hepcidin induction (Figure 2; supplemental Figure 4). However, a more robust hepcidin induction in E coli–infected Hjv−/− mice did not decrease serum iron (Figure 3).
Considering that even Hamp−/− mice respond to LPS by mounting an inadequate hypoferremic response,25 contribution of additional mechanisms26,41,42 is likely. We explored a potential role of ferroportin regulation at the mRNA level. Liver ferroportin mRNA declined dramatically in wild-type or Hjv-deficient mice treated with the (E coli–derived) TLR4 ligand LPS (Figures 1C, 2F, 7G-H; supplemental Figure 4E), infected with E coli (Figure 3F), or treated with the TLR2/6 ligand FSL1 (Figure 3L). The defective hypoferremic response of Hjv-deficient mice, in spite of liver ferroportin mRNA suppression, argues against a major role of ferroportin mRNA regulation in acute hypoferremia of inflammation; nonetheless, a contribution cannot be excluded. Other confounding factors could be reduced dietary iron absorption due to suppression of duodenal iron transporters (Deschemin et al25 ; supplemental Figure 4F-G), as well as increased cellular iron uptake and sequestration within ferritin.43
Hjv-deficient mice exhibit low basal Smad5 phosphorylation in the liver, which underlies the minimal Hamp expression. Nevertheless, they efficiently accumulate phospho-Smad5 following LPS treatment, almost at wild-type levels (Figure 4A). Presumably, this is linked to induction of Inhbb mRNA, which encodes activin B, an upstream activator of Smad signaling.6,7,44 A principal function of activin B in the hepcidin inflammatory pathway was excluded by experiments with Inhbb−/− mice.6 Even though Hjv enhances activin B-dependent hepcidin induction in hepatoma cells and primary hepatocytes,7 it was suggested that activin B–mediated Smad5 phosphorylation in vivo may predominantly occur in nonparenchymal liver cells,6 which exhibit much lower Hjv expression. Our findings are consistent with this view.
Hepatocellular Hjv45 regulates Hamp expression via the Smad pathway by recruiting Bmp ligands to signaling complexes in endosomes.46 The in vitro experiments in Figure 5 demonstrate that Hjv is essential for inflammatory induction of Hamp by recruiting Bmp6, which is secreted from liver sinusoidal endothelial cells.28 Our data are in line with the incapacity of LPS-treated Bmp6−/− mice to activate hepcidin.6 They reinforce the necessity of Smad and Stat cascades for inflammatory induction of hepcidin4,5,47,48 and, furthermore, suggest that the minimal requirement for an effective response is the combination of basal Bmp6/Hjv-dependent Smad and active Il6-dependent Stat signaling.
It should be noted that the specificity of Hjv on BMP6 was evident when primary hepatocytes were exposed to 5 ng/mL of the ligand. This concentration is ∼10 to 25 higher than circulating Bmp6 levels, which are reported to range between 55 and 130 pg/mL in control and iron-loaded mice.49 When hepatocytes were treated with ≥25 ng/mL BMP6, the dependence of Hamp induction on Hjv was partially attenuated (supplemental Figure 7). These findings suggest that pharmacological BMP6 administration could ameliorate hemochromatosis in Hjv−/− mice, as documented in Hfe−/− mice.50 Conceivably, excessive BMP6 may activate Smad signaling upon binding to preformed complexes of type I and II Bmp receptors without the aid of Hjv.46 Such a mechanism would corroborate in vivo evidence that Bmp6 may also operate independently of Hjv.12
Bmp2 is critical for iron homeostasis by regulating hepcidin and exhibits, at least in part, nonoverlapping functions with Bmp6.51 Figure 5 shows that Hjv deficiency only moderately impairs BMP2-mediated Smad signaling and Hamp mRNA induction. The BMP2 dose in this experiment (5 ng/mL) was relatively close to circulating Bmp2 levels (1 ng/mL).52 It is tempting to speculate that Bmp2−/− mice will retain appropriate inflammatory induction of hepcidin following LPS stimulation, contrary to Bmp6−/− counterparts.6
The data in Figures 6 and 7 are in tune with the idea that iron deficiency prevents inflammatory induction of hepcidin by maintaining negligible Bmp6/Smad-signaling activity. Although Bmp6 suppression by iron deficiency contributes to silencing of Smad signaling,33 our results also raise the possibility for direct effects of hepatic iron content on the pathway. Thus, LPS triggered effective inflammatory induction of hepcidin in iron-supplemented mice (Figure 6E-F), in spite of severe Bmp6 mRNA suppression (Figure 7D). Considering that BMPs have a short plasma half-life53 and assuming that circulating Bmp6 levels reflect Bmp6 mRNA, our data suggest that a physiological hepatic iron content is essential for efficient hepcidin induction by iron or inflammatory stimuli.
In conclusion, we demonstrate that Hjv is crucial for hepcidin-mediated hypoferremia in acute inflammation and serves to maintain active Smad signaling via Bmp6. The proposed inflammatory function of Hjv is seemingly contradicted by the dramatic suppression of hepatic Hjv mRNA in LPS-treated mice11,54,55 ; however, this response does not affect steady-state levels of Hjv protein.7 Notably, expression of hepatic Hjv mRNA is only modestly affected in the heat-killed Brucella abortus model of chronic inflammation.7 If Hjv remains stable under chronic inflammatory conditions, it will constitute an attractive pharmacological target against the anemia of inflammation, a frequent morbidity that can be corrected by inhibiting the hepcidin/ferroportin axis.56 In support of this notion, inhibition of Hjv activity using soluble Hjv.Fc47 or anti-Hjv antibodies57 was reported to ameliorate anemia in animal models of anemia of inflammation.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Naciba Benlimame for technical assistance with immunohistochemistry, Jim Gourdon for help with mouse footpad injections, and Charles Dozois (Institut National de la Recherche Scientifique [INRS] Institut Armand-Frappier, Laval, QC, Canada) for providing us E coli SP15.
This work was supported by a grant from the Canadian Institutes of Health Research (CIHR; MOP-86514).
Authorship
Contribution: C.F., N.W., E.C., A.K., and J.W. performed research and analyzed data; and K.P. designed and supervised research and wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Kostas Pantopoulos, Lady Davis Institute for Medical Research, Jewish General Hospital, and Department of Medicine, McGill University, 3755 Cote Ste-Catherine Rd, Montreal, QC H3T 1E2, Canada; e-mail: kostas.pantopoulos@mcgill.ca.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal