

In this issue of Blood, 2 companion papers by 1 and 2 investigate mechanisms underlying immune effector dysfunction in myeloma and highlight T-cell immunoglobulin and ITIM domains (TIGIT) blockade as a novel checkpoint inhibition strategy.
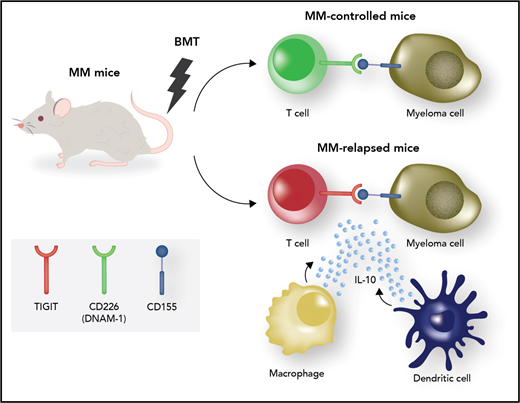
Mechanisms of immune effector dysfunction in myeloma. The myeloma microenvironment in posttransplant relapses is characterized by the presence of TIGIT-expressing dysfunctional T cells displaying attributes of exhaustion (shaded red) as well as interleukin-10 (IL-10)–secreting immature dendritic cells (DCs) and macrophages. T-cell exhaustion can be reversed through blockade of the immune checkpoint TIGIT. The costimulatory counterpart CD226 is expressed by nonexhausted T effectors (shaded green). BMT, bone marrow transplantation; MM, multiple myeloma. Professional illustration by Somersault18:24.
Mechanisms of immune effector dysfunction in myeloma. The myeloma microenvironment in posttransplant relapses is characterized by the presence of TIGIT-expressing dysfunctional T cells displaying attributes of exhaustion (shaded red) as well as interleukin-10 (IL-10)–secreting immature dendritic cells (DCs) and macrophages. T-cell exhaustion can be reversed through blockade of the immune checkpoint TIGIT. The costimulatory counterpart CD226 is expressed by nonexhausted T effectors (shaded green). BMT, bone marrow transplantation; MM, multiple myeloma. Professional illustration by Somersault18:24.
Multiple myeloma,3,4 a tumor of antibody-producing plasma cells, is the most common single blood cancer diagnosis in the United States, with an estimated 30 770 new cases in 2018. Despite the revolution in treatment landscape since the early 2000s, the development of drug resistance remains the almost-universal clinical end point,5 and cures are rare. The “novel agent” era in myeloma therapy saw the introduction of proteasome inhibitors and thalidomide analogs, followed more recently by antibody-based immunotherapies, histone deacetylase inhibitors, and experimental cellular therapies. Autologous transplantation using high-dose melphalan conditioning remains a mainstay of therapy.
Ultimately, the attainment of cures or stable long remissions will require the eradication of every single myeloma cell in the body and/or the development of long-lasting, self-renewing, myeloma antigen–specific central immunological memory. In the early postautologous transplantation period, the often-achieved minimal residual disease state, coupled with immune reconstitution, offers an ideal platform for the administration of immunotherapies that can generate long-term memory. Therefore, the understanding of antimyeloma immune responses and the mechanisms underpinning effector dysfunction are of paramount importance.
In the now-famous chart depicting somatic mutation (and resultant neoantigen) densities across tumor types,6 myeloma occupies a middle-of-the-range position. Myeloma neoantigens provoke immune effector responses that become dysfunctional through bona fide attributes of “exhaustion”7 : cytokine secretion defects, proliferative arrest, and expression of multiple coinhibitory receptors, including TIGIT and programmed cell death protein 1 (PD-1).1,2 The concept of T-cell exhaustion has been challenged by earlier work that characterized T-cell dysfunction in myeloma as telomere-independent immunosenescence.8 Whether through exhaustion or senescence (or a composite state of dysfunction), the myeloma paradigm appears distinct from disseminated hematological malignancies (eg, leukemias), where immunological tolerance appears to result from defective effector priming and anergy.9 Similar to solid tumors, antimyeloma effector responses are efficiently primed but subsequently fail in the battlefield of the tumor bed.
Experience with anti-PD-1 axis checkpoint inhibition in myeloma has been sobering. Single-agent nivolumab was disappointing.10 Combinations of standard antimyeloma agents with pembrolizumab (another anti-PD-1 antibody) were marred by unacceptable toxicity. Therefore, new checkpoint inhibition approaches are urgently needed.
TIGIT is a coinhibitory receptor expressed on the surface of effector T and natural killer (NK) cells, where it competes with its costimulatory counterpart, CD226 (DNAM-1), for binding to the ligands CD155 and CD112 on the surface of myeloma tumor cells (or antigen-presenting cells).11 This paradigm is somewhat reminiscent of antagonistic CTLA-4 and CD28 competing for binding to CD80 and CD86 molecules on the surface of antigen-presenting cells. The Smyth and Martinet research groups showed in earlier work that immunosurveillance in myeloma is dependent on CD226 expressed on the surface of effector T and NK cells.12
In the first paper by Guillerey et al, the authors analyzed CD8+ responses to murine Vκ*MYC myeloma and found that the percentage of CD8+TIGIT+ cells correlated with myeloma burden. Similarly, CD8+ T cells from human patients expressed TIGIT more frequently than other checkpoint molecules. TIGIT+ effector cells from human patients displayed limited cytokine responses to antigen-specific stimulation as well as decreased CD107a, the latter indicating poor killing capability. Vκ*MYC myeloma grew in TIGIT-null mice more slowly, and treatment of wild-type myeloma recipients with anti-TIGIT antibodies reduced tumor burden.
The second study by Minnie et al recapitulated conditions of relapse following stem cell transplant. Chung et al13 had previously shown that in human myeloma, after autologous transplant, dysfunctional T-cells display phenotypic markers of both exhaustion and senescence, and their increase often heralds clinical relapse; these cells express PD-1 and can be reinvigorated to produce effector cytokines following anti-PD-1 treatment ex vivo. In the Minnie et al study, 2 groups of mice emerged posttransplant (see figure): mice with controlled disease and mice with relapsed myeloma. Mechanisms of failure in the latter were further characterized. Relapsed mice showed exhausted CD8+ effectors characterized by TIGIT expression and low CD226. Because IL-10 is known to play a role in T-cell exhaustion, the authors dissected IL-10–dependent mechanisms. Indeed, they found increased numbers of IL-10–producing T cells in relapsed mice. When they transplanted mice with bone marrow cells that were defective in IL-10 production, the clinical outcomes improved as more mice experienced disease control posttransplant. However, in functional experiments, they found that the detrimental IL-10 did not seem to come from, or act upon, T-cells. Rather, the major IL-10 source correlating with failure was myeloid cells, both a somewhat enigmatic DC subset that lacked major histocompatibility complex (MHC) class II expression (but expressed PD-L1) and CD64+ macrophages (see figure). The authors decided to target regulatory macrophages using an anti-CSF1-R antibody and avoid targeting DCs for fear of detrimental effects on effector priming. Indeed, anti-CSF1-R therapy was efficacious. Lastly, in a prelude of what is likely to come in human trials, anti-TIGIT antibody therapy posttransplant resulted in improved rates of disease control.
Several outstanding questions remain. How does myeloid-derived IL-10 cause T-cell dysfunction? Does myeloma tumor cell–derived IL-10 play a role in immune escape? If IL-10 receptor expression on T cells is redundant, does IL-10 signal through another receptor? What is the ontological origin of the immature IL-10–producing DC? Which chemokine networks may be responsible for DC recruitment to the tumor bed? Is myeloma directly preventing DC differentiation or maturation? What is the abundance and activation status of the crucial cross-presenting Batf3-DC subset in patients who progress vs those in remission? How does CSF1-R inhibition impact DC recruitment and differentiation as well as effector function?
The findings presented in the 2 high-impact papers by Guillerey et al and Minnie et al are likely to usher in an era of anti-TIGIT checkpoint inhibition in myeloma. Although some key tiles are still missing from the mechanistic jigsaw puzzle, the picture that emerges is compelling. Myeloma has been known as the hematological malignancy with “carcinoma-like genetics”; intriguingly, it also appears to envy aspects of solid tumor immunobiology. After a bumpy start, checkpoint inhibition in myeloma merits a fresh look.
Conflict-of-interest disclosure: The author declares no competing financial interests.
