

TO THE EDITOR:
Dyskeratosis congenita (DC) and its severe form, Hoyeraal-Hreidarsson syndrome (HHS), are rare and have life-threatening failure of hematopoiesis. Typically, DC patients present with disease features such as nail dystrophy, oral leukoplakia, and abnormal skin pigmentation along with peripheral pancytopenia and marrow hypoplasia with strong predisposition to cancer.1 In DC, hematopoietic failure occurs due to critical shortening of telomeres,2,3 which enhances the DNA damage response4,5 and leads to premature senescence of hematopoietic stem cells.6
Telomeres are lengthened by the enzyme telomerase, which constitutes RNA template (TERC) and the catalytic reverse transcriptase (TERT). Variants affecting telomerase complex (TERT and TERC), telomerase stability (DKC1, NOP10, NHP2, PARN, and NAF1), telomerase trafficking (WRAP53), and telomere replication (CTC1 and RTEL1) have been identified in majority of DC and HHS cases. The shelterin complex proteins (TRF1, TRF2, RAP1, TIN2, POT1, and TPP1) that serve to protect telomeres are critical for telomerase function.7 The first DC variants to be described in a shelterin component were found in TINF2.8 Variants in ACD encoding TPP1 were later described in 2 independent families,9,10 The first was in a family with a history of aplastic anemia, in which the index case had pancytopenia at 8 years of age and was heterozygous for the ACD variant c.499-501del; p.K170 del (K170Δ) that segregated as an autosomal-dominant trait.9 The proband’s mother presented with thrombocytopenia in her 20s and was later diagnosed with myelodysplasia. Her grandmother had mild macrocytic anemia associated with hypocellular bone marrow. The second case, reported by Kocak et al in 2014,10 had several features of HHS, had the same K170Δ variant on one allele along with c.1471 C>T; p.P491T on the other, suggesting an autosomal-recessive inheritance.10 It is of interest that in this family, the proband’s father, who carried the K170Δ variant, had short telomeres but lacked any disease features. Structural and functional studies of K170Δ have not demonstrated a dominant-negative effect on TPP1 function, but instead it is suggested that dosage of TEL patch (patch of amino acids involved in telomerase binding on the surface of TPP1) is responsible for telomere shortening.11,12
The evidence in support of causality in the bone marrow failure (BMF) patients is largely derived from functional analysis of the variants themselves rather than being statistically derived or based on the strength of a significant allelic series.9-11 It remains to be established as to how loss-of-function (LOF) variants in ACD (frameshift, splice donor/acceptor, or stop gain) can have a significant heterozygous frequency in the Genome aggregation database (gnomAD), where 57 of these variants are described (representing ∼6 in 10 000 individuals). Given this frequency of heterozygous LOF ACD variants in the control population, together with the variable pattern of inheritance and very different clinical presentations in the only 2 families described to date, the case for ACD as a proven disease-causing locus remains uncertain.
By whole-exome sequencing in a series of genetically uncharacterized patients (n = 228) presenting with DC or constitutional BMF from our DC registry, we identified nonsynonymous ACD variants in 5 unrelated cases (supplemental Table 1, available on the Blood Web site). In two of these (index cases of families 1 and 2; Figure 1A) the ACD variants were homozygous (supplemental Table 1). The index case of family 1 harbors the homozygous variant c.280C>T; p.V94I, which has been reported once in the homozygous state and 191/270022 alleles in heterozygous state on gnomAD. This index case (age 38 years) had thrombocytopenia, short stature, pulmonary abnormalities, and LSCD (supplemental Table 2; supplemental Figure 1A-B). His older sister, who was heterozygous for this variant, had short stature, pulmonary abnormalities, and LSCD but no hematopoietic defect (supplemental Table 2). The index case from family 2 had a novel homozygous missense variant (c.284T>A, p.L95Q) that has not been reported on gnomAD. The index case, aged 12 years presented with leukoplakia and subsequently developed BMF and immunodeficiency (supplemental Figure 1C-E; supplemental Table 2). His older brother had died of aplastic anemia (supplemental Table 2). No other obvious candidate genes that are previously known to cause BMF are identified in these 2 cases (supplemental Table 3). Both homozygous variants identified in families 1 and 2 are predicted to be damaging by combined annotation dependent depletion score (supplemental Table 1) and affect the highly conserved oligonucleotide-binding (OB)-fold domain in TPP1 that is implicated in telomerase binding, recruitment and function at telomeres (Figure 1B; supplemental Figure 3C).10,12
![Figure 1. TPP1 OB-fold variants cause short telomeres. (A) Family trees showing the segregation of ACD variants. −/−, homozygous variant; +/−, heterozygous; +/+, wild-type (WT). Arrows indicate index cases that underwent whole-exome sequencing. Gray indicates the presence of some somatic features (short stature and limbal stem cell deficiency [LSCD]) but no hematopoietic abnormality. N/A, DNA not available. (B) TPP1 topology showing 3 functional domains: telomerase interacting OB-fold, POT1 recruitment domain (RD), and TIN2 interacting domain (TID) separated by serine threonine (S/T) motif. (C) Whole-blood telomere lengths of index cases and family members are indicated (family 1 in green and family 2 in red). Telomere lengths are reduced in index cases (squares) when compared with heterozygous (diamond) and WT (triangle) family members and controls (n = 218). DAPI, 4′,6-diamidino-2-phenylindole. (D) Telomere fluorescence in situ hybridization of Epstein-Barr virus–transformed lymphoblasts derived from index case in family 2 and a healthy control. (E) Telomere chromatin immunoprecipitation of myc- POT1, FLAG- TPP1 WT, and OB-fold variants expressed in HEK293 cells. Dot blot shows input chromatin used in each immunoprecipitation (top). Immunoprecipitation with an anti-POT1 antibody, anti-FLAG antibody, anti-rabbit (Rb) immunoglobulin G (IgG) control, and protein G beads alone. Expression of TPP1 WT and variants is demonstrated by immunoblotting in the bottom panel. IB, immunoblot. TPP1 p.K170Δ variant was used as positive control, as it has been previously shown to bind to telomeres.9,10 (F) Telomere lengths of S pombe strains harboring Tpz1 (ortholog of human ACD/TPP1) OB-fold variants. Tpz1 K75A yeast strains with previously confirmed short telomeres were used as a positive control.15 Arrow indicates extremely faint telomeric band due to presence of very low telomeric DNA in the L5Q strain, and LC refers to loading control of genomic DNA stained by ethidium bromide.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/132/12/10.1182_blood-2018-03-837799/4/m_blood837799f1.png?Expires=1767744672&Signature=URga4x~6crmI6mCrndfeGjgnUkujF6kwqPKicmCKE7sFEvrPJBe5RlHqqtsHaojoRK3kXvKTDu9~f8Mn1Kjadpp5WRm7PvVBjIXKEZ1J9WYVjArjTnkppBTGlEoXtcHmSp9JYiyoUBoTxomAalBQHYICFUTKtc5Z6gZfsEwMqzA4H9g~zLzvBX4vVgStH9Ksgv7EauovCtFzff1dx58FLEGyqROnTh9Ry3ICLigGYE5vuwlO8X-zKpALzdGWhADJ8t~nOpRjTpR4v42HHKua-jFPBn1wxnDAlxb~tD~Yg-6TlknAd8JUOSqy67bTxeaspzjRxezrTlcMj~F5tcHaYw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
TPP1 OB-fold variants cause short telomeres. (A) Family trees showing the segregation of ACD variants. −/−, homozygous variant; +/−, heterozygous; +/+, wild-type (WT). Arrows indicate index cases that underwent whole-exome sequencing. Gray indicates the presence of some somatic features (short stature and limbal stem cell deficiency [LSCD]) but no hematopoietic abnormality. N/A, DNA not available. (B) TPP1 topology showing 3 functional domains: telomerase interacting OB-fold, POT1 recruitment domain (RD), and TIN2 interacting domain (TID) separated by serine threonine (S/T) motif. (C) Whole-blood telomere lengths of index cases and family members are indicated (family 1 in green and family 2 in red). Telomere lengths are reduced in index cases (squares) when compared with heterozygous (diamond) and WT (triangle) family members and controls (n = 218). DAPI, 4′,6-diamidino-2-phenylindole. (D) Telomere fluorescence in situ hybridization of Epstein-Barr virus–transformed lymphoblasts derived from index case in family 2 and a healthy control. (E) Telomere chromatin immunoprecipitation of myc- POT1, FLAG- TPP1 WT, and OB-fold variants expressed in HEK293 cells. Dot blot shows input chromatin used in each immunoprecipitation (top). Immunoprecipitation with an anti-POT1 antibody, anti-FLAG antibody, anti-rabbit (Rb) immunoglobulin G (IgG) control, and protein G beads alone. Expression of TPP1 WT and variants is demonstrated by immunoblotting in the bottom panel. IB, immunoblot. TPP1 p.K170Δ variant was used as positive control, as it has been previously shown to bind to telomeres.9,10 (F) Telomere lengths of S pombe strains harboring Tpz1 (ortholog of human ACD/TPP1) OB-fold variants. Tpz1 K75A yeast strains with previously confirmed short telomeres were used as a positive control.15 Arrow indicates extremely faint telomeric band due to presence of very low telomeric DNA in the L5Q strain, and LC refers to loading control of genomic DNA stained by ethidium bromide.
TPP1 OB-fold variants cause short telomeres. (A) Family trees showing the segregation of ACD variants. −/−, homozygous variant; +/−, heterozygous; +/+, wild-type (WT). Arrows indicate index cases that underwent whole-exome sequencing. Gray indicates the presence of some somatic features (short stature and limbal stem cell deficiency [LSCD]) but no hematopoietic abnormality. N/A, DNA not available. (B) TPP1 topology showing 3 functional domains: telomerase interacting OB-fold, POT1 recruitment domain (RD), and TIN2 interacting domain (TID) separated by serine threonine (S/T) motif. (C) Whole-blood telomere lengths of index cases and family members are indicated (family 1 in green and family 2 in red). Telomere lengths are reduced in index cases (squares) when compared with heterozygous (diamond) and WT (triangle) family members and controls (n = 218). DAPI, 4′,6-diamidino-2-phenylindole. (D) Telomere fluorescence in situ hybridization of Epstein-Barr virus–transformed lymphoblasts derived from index case in family 2 and a healthy control. (E) Telomere chromatin immunoprecipitation of myc- POT1, FLAG- TPP1 WT, and OB-fold variants expressed in HEK293 cells. Dot blot shows input chromatin used in each immunoprecipitation (top). Immunoprecipitation with an anti-POT1 antibody, anti-FLAG antibody, anti-rabbit (Rb) immunoglobulin G (IgG) control, and protein G beads alone. Expression of TPP1 WT and variants is demonstrated by immunoblotting in the bottom panel. IB, immunoblot. TPP1 p.K170Δ variant was used as positive control, as it has been previously shown to bind to telomeres.9,10 (F) Telomere lengths of S pombe strains harboring Tpz1 (ortholog of human ACD/TPP1) OB-fold variants. Tpz1 K75A yeast strains with previously confirmed short telomeres were used as a positive control.15 Arrow indicates extremely faint telomeric band due to presence of very low telomeric DNA in the L5Q strain, and LC refers to loading control of genomic DNA stained by ethidium bromide.
Telomere length measurement by monochrome multiplex quantitative polymerase chain reaction13 revealed short telomeres in the index case of family 1, just below the tenth centile when compared with his heterozygous sister and controls (n = 218; Figure 1C; supplemental Table 2). Index case from family 2 had very short telomeres, below the first centile as measured by monochrome multiplex quantitative polymerase chain reaction (Figure 1C) and a flow fluorescence in situ hybridization method (supplemental Figure 2). His heterozygous parents also had short telomeres (Figure 1C; supplemental Figure 2; supplemental Table 2) but were asymptomatic. Telomere labeling using a Tel-Cy3 probe (PNA bioscience) on metaphase spreads of Epstein-Barr virus–transformed B lymphoblasts derived from the index case of family 2 (L95Q) revealed reduced telomere signal when compared with an age-matched control (Figure 1D). A telomere chromatin immunoprecipitation assay in TPP1 short hairpin RNA (shRNA)–treated HEK293 cells expressing TPP1-L95Q variant (supplemental Figure 3A-B) revealed no significant change in its association with telomeres (Figure 1E). Furthermore, mimicking TPP1-L95Q variant in Schizosaccharomyces pombe (Tpz1-L5Q) also induced telomere shortening (Figure 1F; supplemental Figure 3C-D).15 These data suggest that the TPP1-L95Q OB-fold variant interferes with telomere maintenance despite localizing to telomeres.
The TEL patch in the OB-fold domain is essential for telomerase recruitment at telomeres (Figure 2A).12,16 TPP1 OB-fold crystal structure reveals both V94 and L95 residues that are mutated in our cases are present in the N terminus of OB-fold domain, forming a hydrophobic cleft that extends the TEL patch (Figure 2A).12,14 Mutating both these residues simultaneously reduces telomerase processivity, abrogates telomerase recruitment to telomeres, and shortens telomeres of human cells in culture.14 In line with this, coimmunoprecipitation and quantitative TRAP analysis in HeLa nuclear lysates demonstrated that when overexpressed, both TPP1-V94I and TPP1-L95Q have impaired ability to bind TERT (Figure 2B-C) and reduced TPP1 associated telomerase activity in comparison with WT (Figure 2D-E). These results indicate that both OB-fold residues V94 and L95 participate in TPP1–telomerase interaction and regulate telomerase activity. Telomerase association with TPP1 OB-fold, is important for its maturation, traffic and recruitment to telomeres via Cajal bodies.17 In HeLa cells, when overexpressed, TPP1-L95Q showed diffused punctate in the nucleoplasm that did not colocalize with coilin that resides in Cajal bodies (Figure 2F-G). This is similar for TPPI-K170Δ and other artificial TEL patch variants described previously.9-12,14 Interestingly, abnormal cytoplasmic staining of TPP1-L95Q (Figure 2H-I) and reduction in telomerase activity were observed in the patient cells (Figure 2J), indicating defects in the telomerase maturation pathway.
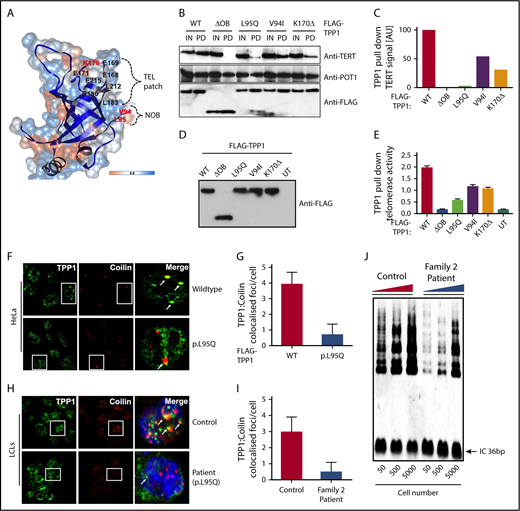
Characterization of TPP1 OB-fold variants extend TEL patch functional domain. (A) In silico model of the TPP1 OB-fold crystal structure (Protein Data Bank accession number 2I46)16 using UCSF CHIMERA, an extensible molecular modeling software. Previously described TPP1-TEL patch residues are denoted in black,12 and the patient variants in BMF patients identified to date are denoted in red. OB-fold ribbon structure is depicted in blue, and surface hydrophobicity preset is shown with amino acid hydrophobicity in the Kyte-Doolittle scale with colors ranging from dodger blue for the most hydrophilic to white at 0.0 to orange for the most hydrophobic. NOB refers to the N terminus of the OB-fold domain that is recently described in telomerase binding.14 (B-C) Immunoblotting of FLAG pull-down (PD) complexes from nuclear extracts of HEK293 cells that are treated with TPP1 3′ untranslated region shRNA and expressing shRNA-resistant FLAG-TPP1 variants along with TERT and POT1. IN refers to 15% input. Note the reduction in TERT signal pulled by the TPP1 OB-fold variants compared with input and WT. (D-E) HeLa cells expressing FLAG-tagged WT TPP1 were analyzed for TPP1-associated endogenous telomerase activity by immunoprecipitation and subsequent telomerase repeated amplification (TRAP) analysis. Results from TRAP assay were normalized based on the amount of eluted TPP1 protein. (F-G) Double immunofluorescence was used to detect the FLAG-TPP1 proteins (green) and coilin (a Cajal body marker, red) in HeLa cells. Yellow (merged green + red) spots (indicated by arrows) show colocalization of TPP1 with coilin, which was quantified by counting cells from different fields of view (n = 2). (H-I) Endogenous TPP1 colocalization with coilin in control and patient (TPP1 p.L95Q) B lymphoblastoid cells. (J) Relative levels of telomerase activity in patient and age-matched control B lymphoblastoid cells at passage 5 were determined by TRAP assay.
Characterization of TPP1 OB-fold variants extend TEL patch functional domain. (A) In silico model of the TPP1 OB-fold crystal structure (Protein Data Bank accession number 2I46)16 using UCSF CHIMERA, an extensible molecular modeling software. Previously described TPP1-TEL patch residues are denoted in black,12 and the patient variants in BMF patients identified to date are denoted in red. OB-fold ribbon structure is depicted in blue, and surface hydrophobicity preset is shown with amino acid hydrophobicity in the Kyte-Doolittle scale with colors ranging from dodger blue for the most hydrophilic to white at 0.0 to orange for the most hydrophobic. NOB refers to the N terminus of the OB-fold domain that is recently described in telomerase binding.14 (B-C) Immunoblotting of FLAG pull-down (PD) complexes from nuclear extracts of HEK293 cells that are treated with TPP1 3′ untranslated region shRNA and expressing shRNA-resistant FLAG-TPP1 variants along with TERT and POT1. IN refers to 15% input. Note the reduction in TERT signal pulled by the TPP1 OB-fold variants compared with input and WT. (D-E) HeLa cells expressing FLAG-tagged WT TPP1 were analyzed for TPP1-associated endogenous telomerase activity by immunoprecipitation and subsequent telomerase repeated amplification (TRAP) analysis. Results from TRAP assay were normalized based on the amount of eluted TPP1 protein. (F-G) Double immunofluorescence was used to detect the FLAG-TPP1 proteins (green) and coilin (a Cajal body marker, red) in HeLa cells. Yellow (merged green + red) spots (indicated by arrows) show colocalization of TPP1 with coilin, which was quantified by counting cells from different fields of view (n = 2). (H-I) Endogenous TPP1 colocalization with coilin in control and patient (TPP1 p.L95Q) B lymphoblastoid cells. (J) Relative levels of telomerase activity in patient and age-matched control B lymphoblastoid cells at passage 5 were determined by TRAP assay.
In summary, we report the first instance of homozygous TPP1 OB-fold variants in patients from 2 unrelated families who have overlapping phenotypes with DC.18 As these variants cluster in the TEL patch of TPP1, which is essential for telomerase recruitment to telomeres, they provide in vivo evidence in humans for the biological importance of this TPP1 OB-fold domain.
The online version of this article contains a data supplement.
Acknowledgments
The authors thank all the families and clinicians who contributed to this research and the staff at Genome Centre Blizard Institute for help with DNA sequencing.
This work was supported by the Medical Research Council (grant MR/P018440/1), Bloodwise (grant 14032), and Children with Cancer UK (grant 2013/144).
Authorship
Contribution: T.V. and I.D. are the principal investigators; H.T. and L.C.C. performed most of the experiments; A.J.W., A.E., Z.S., N.P., J.F., K.T., and S.C. contributed to the experiments. N.Y., T.A., D.A., R.F.A., and J.T. performed clinical analysis; and H.T., T.V., and I.D. wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Hemanth Tummala, Blizard Institute, Barts and The London School of Medicine and Dentistry, Queen Mary University of London, Newark St, London E12AT, United Kingdom; e-mail: h.tummala@qmul.ac.uk.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal