

Key Points
Alterations in JAK/STAT signaling pathway are highly prevalent in PTCL and NKTL, where STAT3 and TP53 are the most frequently mutated genes.
STAT3 activation drives PD-L1 expression in NKTL, providing a rationale to combine STAT3 inhibitors with immune checkpoint inhibitors.

Abstract
Mature T-cell lymphomas, including peripheral T-cell lymphoma (PTCL) and extranodal NK/T-cell lymphoma (NKTL), represent a heterogeneous group of non-Hodgkin lymphomas with dismal outcomes and limited treatment options. To determine the extent of involvement of the JAK/STAT pathway in this malignancy, we performed targeted capture sequencing of 188 genes in this pathway in 171 PTCL and NKTL cases. A total of 272 nonsynonymous somatic mutations in 101 genes were identified in 73% of the samples, including 258 single-nucleotide variants and 14 insertions or deletions. Recurrent mutations were most frequently located in STAT3 and TP53 (15%), followed by JAK3 and JAK1 (6%) and SOCS1 (4%). A high prevalence of STAT3 mutation (21%) was observed specifically in NKTL. Novel STAT3 mutations (p.D427H, E616G, p.E616K, and p.E696K) were shown to increase STAT3 phosphorylation and transcriptional activity of STAT3 in the absence of cytokine, in which p.E616K induced programmed cell death-ligand 1 (PD-L1) expression by robust binding of activated STAT3 to the PD-L1 gene promoter. Consistent with these findings, PD-L1 was overexpressed in NKTL cell lines harboring hotspot STAT3 mutations, and similar findings were observed by the overexpression of p.E616K and p.E616G in the STAT3 wild-type NKTL cell line. Conversely, STAT3 silencing and inhibition decreased PD-L1 expression in STAT3 mutant NKTL cell lines. In NKTL tumors, STAT3 activation correlated significantly with PD-L1 expression. We demonstrated that STAT3 activation confers high PD-L1 expression, which may promote tumor immune evasion. The combination of PD-1/PD-L1 antibodies and STAT3 inhibitors might be a promising therapeutic approach for NKTL, and possibly PTCL.
Introduction
Mature T-cell lymphomas, including peripheral T-cell lymphoma (PTCL) and NK/T-cell lymphoma (NKTL), appear to have a geographical predilection for Asia.1-3 The World Health Organization classification recognizes a number of distinctive subtypes of PTCL and NKTL, including angioimmunoblastic T-cell lymphoma, anaplastic lymphoma kinase-positive (ALK+) and anaplastic lymphoma kinase-negative (ALK−) anaplastic large cell lymphoma (ALCL), cutaneous T-cell lymphoma (CTCL), and PTCL not otherwise specified (PTCL-NOS).4 With the exception of ALK+ ALCL, patients with PTCL and NKTL generally have a poor prognosis, with 5-year overall survival rates less than 40%.4
Multiple studies have suggested that the JAK/STAT pathway plays a significant role in the pathogenesis of PTCL and NKTL. We previously identified JAK3 activating mutations in about one-third of NKTL cases.5 Activating mutations of JAK1 and/or STAT3 were found in 18% of ALK− ALCL, while being absent either in ALK+ ALCL or in PTCL-NOS.6 In contrast, JAK1 and STAT5B mutations were recently reported in 2 of 4 patients with PTCL-NOS.7 Collectively, the mutation frequencies varied greatly among the PTCL and NKTL subtypes and between studies. In NKTL, JAK3 was constitutively activated in 87% of cases; however, only 21% of these could be explained by mutations. This suggests the presence of key activating and/or cooperating mutations other than the JAKs and STATs.8
Programmed cell death-ligand 1 (PD-L1) and programmed cell death-1 (PD-1) are important immune checkpoint molecules involved in immune evasion.9 PD-1/PD-L1 blockade by monoclonal antibodies has achieved great efficacy and is approved by the US Food and Drug Administration for a number of malignancies such as gastric carcinoma,10 urothelial carcinoma,11 melanoma,12 and classical Hodgkin lymphoma (cHL).13 Recently, PD-1 blockade demonstrated a promising clinical response in patients with relapsed or refractory NKTL, and a strong PD-L1 expression level was found to correlate with better outcome.14,15 The genetic and molecular basis of PD-L1 overexpression has been investigated in multiple hematological malignancies.16-18 In cHL, copy gains of 9p24.1 locus resulted in PD-L1 overexpression,16 and in diffuse large B-cell lymphoma and adult T-cell lymphoma, the structural variants disrupting the 3′ region of the PD-L1 gene led to aberrant transcripts with elevated expression.19 In ALK+ ALCL, the NPM-ALK oncogenic fusion induced the expression of PD-L1 through STAT3,20 whereas in ALK− ALCL, STAT3 and MYC transcriptionally upregulated PD-L1,18 highlighting the interaction between the JAK/STAT and PD-1/PD-L1 pathways. Whether such an interaction exists in subtypes of PTCL and NKTL other than ALCL remains to be explored.
In this study, we determined the prevalence of JAK/STAT pathway alterations in both PTCL and NKTL, using a targeted deep sequencing approach with the aim of identifying potential therapeutic targets. We performed detailed functional and structural characterization of novel STAT3-activating mutations and demonstrated a regulatory role of STAT3 activation in PD-L1 expression. Targeting STAT3 might have a synergistic effect in immune checkpoint blockade therapy.
Methods
Patients and cell lines
One hundred seventy-one PTCL and NKTL samples were collected from Singapore General Hospital and National University Hospital in Singapore and Guangdong General Hospital and Sun Yat-sen University Cancer Center in China; all patients provided written informed consent. NKTL was the most common subtype (101 tumor tissues, 8 cell lines), followed by ALCL (12 ALK+ ALCL, 13 ALK− ALCL, and 2 cutaneous ALCL tumor tissues), PTCL-NOS (26 tumor tissues), and CTCL (8 tumor tissues, 1 cell line). Peripheral blood or buccal swabs of 35 matched normal control patients were included. The clinical and pathological characteristics of our study subjects are summarized in supplemental Table 1, available on the Blood Web site. Cell lines and cell culture conditions are described in supplemental Methods. This study was approved by the SingHealth Centralized Institutional Review Board (study number 2004/407/F).
Primary natural killer cell isolation for western blot
Primary human natural killer (NK) cells were isolated from peripheral blood mononuclear cells by depletion of non-NK cells using a human NK cell isolation kit (Miltenyi Biotec). Purity of NK cells was evaluated by CD56-PE staining, and samples with more than 90% CD56+ cells were used. Cells were cultured in X-VIVO 15 medium (Lonza) supplemented with 5% human serum (Innova Biosciences) with 200 U/mL interleukin 2 (IL-2; Proleukin).
Genomic DNA extraction
Deep-targeted capture sequencing
Targeted capture sequencing was performed with a customized capture probe set that targeted exons of 188 JAK/STAT pathway-related genes (supplemental Table 2). One hundred seventy-one PTCL and NKTL gDNA samples were sequenced on the HiSeq2000 platform (Illumina) to a mean depth of 726-fold (supplemental Table 3). GATK was used to call single-nucleotide substitutions and insertion/deletions (indels), and candidate variants were annotated using wAnnovar. A subset of mutations was randomly selected and verified by Sanger sequencing, as described previously,5 with a validation rate of 72%. Primer sequences are listed in supplemental Table 4, and detailed methods are described in supplemental Methods. Sequencing data are available on the European Genome-phenome Archive (accession number: EGAS00001002740).
Generation and expression of STAT3 constructs
Wild-type STAT3 (STAT3WT) was amplified with Q5 High-Fidelity DNA Polymerase (NEB), using NK-S1 cell line22 cDNA as the template, and cloned into retroviral plasmid pMIGR1 (Addgene; plasmid no. 27490), using XhoI and HpaI restriction sites. STAT3 mutations were generated from STAT3WT constructs, using QuikChange II XL Site-Directed Mutagenesis Kit (Agilent Technologies), and confirmed by Sanger sequencing. Retroviral vectors harboring STAT3WT and STAT3 mutations were introduced into Ba/F3 and NK-S1 cells to develop stable cell lines. Transduced cells were selected on the basis of green fluorescent protein positivity, using BD Aria III Cell Sorter (BD Biosciences).
Cell viability assays
For IL-3 independence assays, Ba/F3 cells were treated with 0 and 10 ng/mL IL-3 at day (D)0, D1, D2, and D3. For Stattic treatment assays, Ba/F3 and NK-S1 cells were treated with dimethyl sulfoxide and Stattic at indicated concentrations for 72 hours. Cell viability was determined using CellTiter-Glo Luminescent Cell Viability Assay (Promega). Dose–response curves were plotted and 50% inhibitory concentration was calculated using GraphPad Prism.
GapmeR-mediated knockdown of STAT3
To knockdown STAT3 in NKTL cell lines, we designed specific GapmeR targeted against STAT3 and applied as described previously,23 with minor modifications. Briefly, 1 × 106 cells were nucleofected with 10 nM GapmeR, using a 4D-Nucleofectorsystem (Lonza) and Amaxa Nucleofector Kit. An equivalent amount of nontargeting GapmeR was used as a control. After GapmeR nucleofection, cells were incubated for 72 hours and analyzed.
Immunohistochemistry
Immunohistochemistry staining in formalin-fixed, paraffin-embedded tissues of patients with NKTL for phosphorylated STAT3 (pSTAT3) and PD-L1 were performed as previously described,24 with pSTAT3 rabbit monoclonal antibody (D3A7; Cell Signaling Technology) and anti-PD-L1 rabbit monoclonal antibody (SP263; Ventana), respectively. Scoring of pSTAT3 and PD-L1 expression is described in supplemental Methods.
Western blot, real-time quantitative polymerase chain reaction and whole-transcriptome sequencing
Experimental procedures for western blot and real-time quantitative polymerase chain reaction (RT-qPCR) were performed as previously described,21 and methods for whole-transcriptome sequencing are described in supplemental Methods. Antibodies and primers are listed in supplemental Tables 5 and 6, respectively.
STAT3 chromatin immunoprecipitation qPCR
Chromatin immunoprecipitation was conducted as previously described,25 using anti-STAT3 mouse monoclonal antibody (124H6; Cell Signaling Technology). Five percent of the reaction was removed as input chromatin. Primer sets were designed to amplify 2 sites within the mouse PD-L1 promoter region and a negative control region outside the promoter region, which are listed in supplemental Table 7. Enrichment data were analyzed by calculating the immunoprecipitated DNA as a percentage of input DNA.
Flow cytometry
Ba/F3 cells were stained with mouse PD-L1 antibody (2096C; R&D Systems) and rabbit immunoglobulin G isotype control (Invitrogen). NKTL cells were stained with human PD-L1 antibody (MIH1; BD Pharmingen) and PE-conjugated mouse immunoglobulin G1 κ isotype control (eBioscience). The stained cells were analyzed by LSRII (BD Bioscience) and FACS Canto II (BD Bioscience), and quantified using FlowJo (v7.2.2). Mean fluorescence intensity (MFI) was calculated by subtracting the isotype control MFI from PD-L1 MFI.
STAT3 modeling
Statistical analyses
Continuous variables were compared using a 2-tailed Student t test, and categorical variables were compared using Fisher’s exact test. Statistical significance was reached with a P value < .05. Statistical analyses were performed using SPSS version 18.0.
Results
Somatic alteration of JAK/STAT pathway is highly prevalent in PTCL and NKTL
To determine the prevalence of JAK/STAT pathway alteration in PTCL and NKTL, we performed targeted capture sequencing for 188 genes associated with this oncogenic pathway in 171 cases. A total of 272 somatic mutations in 101 genes were identified in 73% (125/171) of samples (supplemental Table 8). The variants comprised 245 missense, 12 nonsense, and 1 stop loss single-nucleotide substitutions and 7 frameshift and 7 inframe coding indels. Alterations of JAK/STAT genes were observed across all subtypes (78% in NKTL [85/109], 67% in ALCL [18/27], 33% in CTCL [3/9], and 54% in PTCL-NOS [14/26]), suggesting that the JAK/STAT pathway is frequently altered in PTCL and NKTL.
A total of 52 genes were found recurrently mutated (supplemental Table 9), where STAT3 and tumor protein P53 (TP53) were most frequently mutated, followed by JAK3, JAK1, and suppressor of cytokine signaling 1 (SOCS-1; Figure 1A). STAT3 mutations were observed in 15% of cases (25/171), with 52% (13/25) carrying known hotspot-activating mutations (p.D661Y in 4 cases; p.G618R in 3 cases; p.S614R, p.Y640F, and p.N647I each in 2 cases) located within the Src homology 2 (SH2) domain, which mediates the dimerization and activation of STAT protein28 (Figure 1B). A total of 8 novel STAT3 missense mutations located in the coiled coil (p.D171N), DNA-binding (p.D427H, p.D566N), and SH2 (p.E616G, p.E616K, p.V667L, p.E696K, p.P715L) domains were identified in our study. TP53 was mutated in 15% of cases (25/171), resulting in 1 deletion and 20 missense and 4 nonsense mutations. Recurrent mutations (p.C96F, p.123IN, p.G206D, and p.R209Q) found in the COSMIC database and located within the DNA binding domain were observed in 9 cases.29 We identified 3 samples carrying 2 separate TP53 mutations, suggesting total loss of TP53 function. Interestingly, STAT3 mutation frequency was significantly associated with subtype, where STAT3 was more frequently mutated in NKTL than ALCL and PTCL-NOS, but not CTCL (NKTL vs ALCL, P = .045; NKTL vs PTCL-NOS, P = .045; NKTL vs CTCL, P = .20; pairwise comparison using Fisher’s exact test), whereas TP53 was primarily seen in PTCL-NOS. This indicates that although the JAK/STAT pathway is highly altered in PTCL and NKTL, certain genes within the pathway are selectively mutated within each subtype (Table 1; supplemental Figure 1A-D).

Frequently mutated genes in the JAK/STAT signaling pathway identified by targeted capture sequencing in PTCLs. (A) Distribution of mutations across PTCL subtypes (ALCL, CTCL, NKTL, and PTCL-NOS). The top 36 most frequently mutated genes are shown with the last row indicating the first gene with 2 recurrent mutations. The bar on the right represents the number of samples with mutations, and the bar on the top represents the number of mutations in each sample. Samples having no mutations were excluded. (B) Locations of novel, previously studied, and hotspot mutations in the coiled-coil α domain, DNA binding domain and SH2-domain of STAT3 in PTCLs. STAT3 mutations p.R278H,79 p.H410R,63 and p.Q344H80 were previously reported in autoimmune lymphoproliferative syndrome, large granular lymphocyte leukemia, and patients with lymphoproliferation and childhood-onset autoimmunity, respectively.
Frequently mutated genes in the JAK/STAT signaling pathway identified by targeted capture sequencing in PTCLs. (A) Distribution of mutations across PTCL subtypes (ALCL, CTCL, NKTL, and PTCL-NOS). The top 36 most frequently mutated genes are shown with the last row indicating the first gene with 2 recurrent mutations. The bar on the right represents the number of samples with mutations, and the bar on the top represents the number of mutations in each sample. Samples having no mutations were excluded. (B) Locations of novel, previously studied, and hotspot mutations in the coiled-coil α domain, DNA binding domain and SH2-domain of STAT3 in PTCLs. STAT3 mutations p.R278H,79 p.H410R,63 and p.Q344H80 were previously reported in autoimmune lymphoproliferative syndrome, large granular lymphocyte leukemia, and patients with lymphoproliferation and childhood-onset autoimmunity, respectively.
Comparison of mutational frequencies of genes among PTCL and NKTL, using Fisher's exact test (4 × 2 contingency table)
| Gene | NKTL | ALCL | PTCL-NOS | CTCL | P | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mutant (N) | Wild-type (N) | Proportion of Mutants (%) | Mutant (N) | Wild-type (N) | Proportion of Mutants (%) | Mutant (N) | Wild-type (N) | Proportion of Mutants (%) | Mutant (N) | Wild-type (N) | Proportion of Mutants (%) | ||
| STAT3 | 23 | 86 | 21.1 | 1 | 26 | 3.7 | 1 | 25 | 3.8 | 0 | 9 | 0.0 | .018 |
| TP53 | 13 | 96 | 11.9 | 4 | 23 | 14.8 | 7 | 19 | 26.9 | 1 | 8 | 11.1 | .264 |
| JAK3 | 9 | 100 | 8.3 | 0 | 27 | 0.0 | 1 | 25 | 3.8 | 1 | 8 | 11.1 | .335 |
| JAK1 | 7 | 103 | 6.4 | 2 | 25 | 7.4 | 0 | 26 | 0.0 | 1 | 8 | 11.1 | .407 |
| SOCS1 | 4 | 105 | 3.7 | 0 | 27 | 0.0 | 2 | 24 | 7.7 | 0 | 9 | 0.0 | .439 |
| Gene | NKTL | ALCL | PTCL-NOS | CTCL | P | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mutant (N) | Wild-type (N) | Proportion of Mutants (%) | Mutant (N) | Wild-type (N) | Proportion of Mutants (%) | Mutant (N) | Wild-type (N) | Proportion of Mutants (%) | Mutant (N) | Wild-type (N) | Proportion of Mutants (%) | ||
| STAT3 | 23 | 86 | 21.1 | 1 | 26 | 3.7 | 1 | 25 | 3.8 | 0 | 9 | 0.0 | .018 |
| TP53 | 13 | 96 | 11.9 | 4 | 23 | 14.8 | 7 | 19 | 26.9 | 1 | 8 | 11.1 | .264 |
| JAK3 | 9 | 100 | 8.3 | 0 | 27 | 0.0 | 1 | 25 | 3.8 | 1 | 8 | 11.1 | .335 |
| JAK1 | 7 | 103 | 6.4 | 2 | 25 | 7.4 | 0 | 26 | 0.0 | 1 | 8 | 11.1 | .407 |
| SOCS1 | 4 | 105 | 3.7 | 0 | 27 | 0.0 | 2 | 24 | 7.7 | 0 | 9 | 0.0 | .439 |
JAK3 mutations were detected in 6% of samples (11/171). Three major hotspots were identified, p.M511I, p.A572V, and p.A573V, which are located in the pseudokinase domain region of JAK3 and have been shown to confer gain of function.30-32 A single case was found to carry 2 separate JAK3 mutations (p.M511I and p.L575F), indicating profound alteration of JAK3 function. JAK1 was mutated in 6% of cases (10/171), and the mutations identified mostly affect the 652nd codon (p.652D/H) within the pseudokinase domain and the 1097th codon (p.G1097D/V) within the kinase domain of JAK1. Mutations in SOCS-1, an inhibitor of JAK/STAT signaling, were detected in 4% of samples (6/171), resulting in 1 nonsense mutation (p.C146X) and 5 missense mutations (p.A89P, p.T100I, p.F144L, p.M161I, and p.P165L). These alterations that occur within the SH2 domain required for JAK binding and inhibition of JAK/STAT signaling could potentially lead to loss of SOCS-1 function.33 Prevalence of JAK3 and JAK1 alterations appeared highest in CTCL, whereas SOCS-1 alteration appeared highest in PTCL-NOS; however, differences did not reach statistical significance among the subtypes, which could be attributed to the small sample size of CTCL (Table 1).
Collectively, mutations in the JAK/STAT genes were not mutually exclusive from each other (supplemental Table 10). It is remarkable that 62% (106/171) of cases displayed co-occurring mutations in at least 2 members of the JAK/STAT pathway, a phenomenon reported in previous studies.6,7 The coexistence of JAK1/STAT3 and JAK3/STAT5B mutations was reported in ALK− ALCL6 and primary intestinal T-cell lymphomas,7,21 respectively, suggesting that double mutants could act synergistically to activate the JAK/STAT pathway.
Novel STAT3 mutations are activating and sensitive to pharmacologic inhibition
STAT3 mutation is the most frequently mutated gene in NKTL (Table 1; supplemental Figure 1C). Among the 8 novel STAT3 mutations, 4 mutations (p.D427H, p.E616G, p.E616K, and p.E696K) were predicted by FATHMM to be damaging (supplemental Table 8), and their amino acid mutations involved a charged or polar group.34 To explore the functional implications of these mutations, we generated expression constructs for wild-type, p.D427H, p.E616G, p.E616K, and p.E696K variants of the STAT3 protein. When expressed in IL-3-dependent murine lymphoid Ba/F3 cells, immunoblot analysis (Figure 2A) demonstrated increased autophosphorylation of STAT3 at Tyr705 residue in STAT3 mutants, but not in STAT3WT and empty vector in the absence of IL-3, where different levels of pSTAT3 were observed for different mutants. Furthermore, the gene expression of downstream targets of STAT3 such as c-myc,35 survivin,36 and HIF1A37 were upregulated by STAT3 mutants when compared with STAT3WT or empty vector, suggesting activation of the downstream signaling cascade (Figure 2B). However, among the STAT3 mutants, only pE616K mutant conferred IL-3-independent growth to Ba/F3 cells (Figure 2C). When Ba/F3 cells were treated with increasing concentrations of STAT3 inhibitor, Stattic, in the presence of IL-3, STAT3 mutants were found to be more sensitive to Stattic compared with STAT3WT and empty vector (Figure 2D), which was also observed in p.E616K-expressing NK-S1 cells, a cytokine-independent NKTL cell line (supplemental Figure 2). Together, these findings demonstrated that novel STAT3 mutations (p.D427H, p.E616G, p.E616K, and p.E696K) resulted in constitutive activation of STAT3 and are sensitive to pharmacologic inhibition.
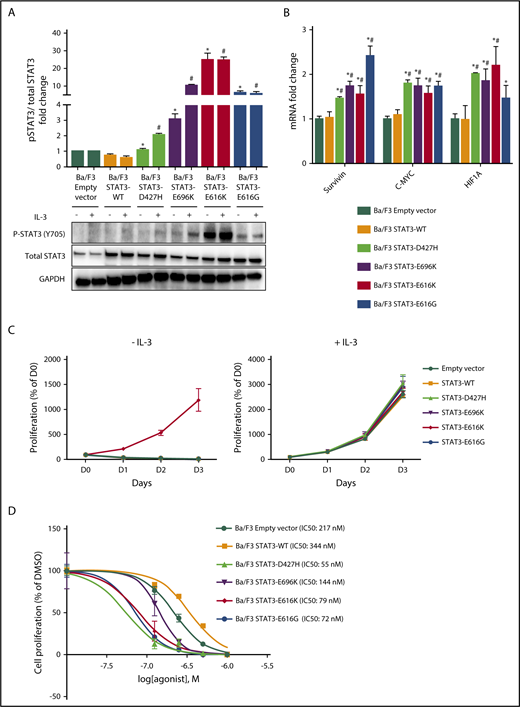
Novel STAT3 mutations cause constitutive STAT3 activity and are sensitive to pharmacologic inhibition. (A) Western blot analysis of pSTAT3 (Y705) and total STAT3 protein expression level in Ba/F3 cells expressing empty vector, STAT3WT, and novel STAT3 mutants after culture for 6 hours in medium with and without IL-3. Bands were quantified with Image J, and protein expression levels were represented as fold change of pSTAT3/total STAT3 relative to empty vector. *P < .05 compared with STAT3WT in medium without IL-3; #P < .05 compared with STAT3WT in medium with IL-3. (B) mRNA expression of STAT3 target genes in Ba/F3 cells expressing empty vector, STAT3WT, and novel STAT3 mutants after culture for 6 hours in medium without IL-3. Results were represented as fold change relative to empty vector and normalized against housekeeping gene NONO. *P < .05 compared with empty vector; #P < .05 compared with STAT3WT. (C) Cell viability assays of Ba/F3 cells expressing empty vector, STAT3WT, and novel STAT3 mutants with and without IL-3 up to 72 hours. (D) Cell viability assays with dimethyl sulfoxide vehicle and Stattic (0.125 µM, 0.25 µM, 0.5 µM, and 1.0 µM) for 72 hours in empty vector, STAT3WT, and novel STAT3 mutant Ba/F3 cells. All results are expressed as mean ± SD of 3 independent experiments.
Novel STAT3 mutations cause constitutive STAT3 activity and are sensitive to pharmacologic inhibition. (A) Western blot analysis of pSTAT3 (Y705) and total STAT3 protein expression level in Ba/F3 cells expressing empty vector, STAT3WT, and novel STAT3 mutants after culture for 6 hours in medium with and without IL-3. Bands were quantified with Image J, and protein expression levels were represented as fold change of pSTAT3/total STAT3 relative to empty vector. *P < .05 compared with STAT3WT in medium without IL-3; #P < .05 compared with STAT3WT in medium with IL-3. (B) mRNA expression of STAT3 target genes in Ba/F3 cells expressing empty vector, STAT3WT, and novel STAT3 mutants after culture for 6 hours in medium without IL-3. Results were represented as fold change relative to empty vector and normalized against housekeeping gene NONO. *P < .05 compared with empty vector; #P < .05 compared with STAT3WT. (C) Cell viability assays of Ba/F3 cells expressing empty vector, STAT3WT, and novel STAT3 mutants with and without IL-3 up to 72 hours. (D) Cell viability assays with dimethyl sulfoxide vehicle and Stattic (0.125 µM, 0.25 µM, 0.5 µM, and 1.0 µM) for 72 hours in empty vector, STAT3WT, and novel STAT3 mutant Ba/F3 cells. All results are expressed as mean ± SD of 3 independent experiments.
Modeling of functional STAT3 mutations
Analysis of the STAT3 homodimer structural model (supplemental Figure 3) showed that the residues p.D427 and p.E616 are positioned at the symmetric dimerization interface of the SH2 domain, in which mutations (p.D427H, p.E616G, and p.E616K) could confer higher polarity or hydrophobicity to the SH2 dimerization surface, potentially modulating the affinity of reciprocal phosphotyrosine-SH2 interactions and increasing stabilization of STAT3 homo- or heterodimers, and thus the activation of STAT3.28,38 Although the p.E696 residue is not involved in the SH2 dimerization interface, it is near the transcription activation domain involved in the recruitment of transcriptional activators.39 Modifications in this residue could therefore increase the transcriptional activity of STAT3.
STAT3 activation directly drives PD-L1 expression in NKTL
A recent report suggested that the high response rate of patients with relapse/refractory NKTL to immune checkpoint blockade could be contributed by the high PD-L1 expression in the tumors.14 To determine the relationship between STAT3 activity and PD-L1 expression, a panel of NKTL cell lines with established genetic information was used. NKTL cell lines harboring STAT3 mutations such as SNT8 (p.D661Y), SNK6 (p.D661Y), and NKYS (p.Y640F) displayed constitutive STAT3 phosphorylation at the Tyr705 residue with a concomitant high level of PD-L1 expression (Figure 3A-C), suggesting that these STAT3 mutations might be involved in driving PD-L1 expression. Importantly, expression of novel STAT3 mutations (p.E616K and p.E616G) in NK-S1, a STAT3WT NKTL cell line, increased STAT3 activation and induced PD-L1 expression (Figure 3D-F), which was further confirmed in Ba/F3 cells (supplemental Figure 4A-C). Reciprocally, depletion of STAT3 using STAT3 Gapmer, a locked nucleic acid-conjugated chimeric single strand antisense oligonucleotide in NKYS and SNK6, diminished expression of PD-L1 (Figure 4A-C). Consistently, treatment with a STAT3 inhibitor, Stattic, in NKYS and SNK6 reduced pSTAT3 and PD-L1 expression levels (Figure 4D-F), which was also observed in p.E616K-expressing Ba/F3 cells (supplemental Figure 5). These results suggest a strong association between STAT3 activity and PD-L1 expression.

Expression of PD-L1 is induced by STAT3 in NKTL. (A) Protein expression of pSTAT3 (Y705), total STAT3, and PD-L1 examined by western blot in NKTL cell lines at basal state. (B) Expression of PD-L1 mRNA examined by RT-qPCR in NKTL cell lines at basal state, and results were normalized against housekeeping gene CHMP2A. (C) Expression of membranous PD-L1 examined by flow cytometry in NKTL cell lines at basal state. (D) NK-S1 cells were transduced with empty vector, STAT3WT, p.E616K, and p.E616G expression vectors. The pSTAT3 and total STAT3 protein levels in these cells were detected with western blot. (E) PD-L1 mRNA in these cells was detected by RT-qPCR. Results were represented as fold change relative to empty vector and normalized against housekeeping gene CHMP2A. (F) Membranous PD-L1 expression in these cells was detected by flow cytometry. All results are expressed as mean ± SD of 3 independent experiments. *P < .05 compared with empty vector; #P < .05 compared with STAT3WT.
Expression of PD-L1 is induced by STAT3 in NKTL. (A) Protein expression of pSTAT3 (Y705), total STAT3, and PD-L1 examined by western blot in NKTL cell lines at basal state. (B) Expression of PD-L1 mRNA examined by RT-qPCR in NKTL cell lines at basal state, and results were normalized against housekeeping gene CHMP2A. (C) Expression of membranous PD-L1 examined by flow cytometry in NKTL cell lines at basal state. (D) NK-S1 cells were transduced with empty vector, STAT3WT, p.E616K, and p.E616G expression vectors. The pSTAT3 and total STAT3 protein levels in these cells were detected with western blot. (E) PD-L1 mRNA in these cells was detected by RT-qPCR. Results were represented as fold change relative to empty vector and normalized against housekeeping gene CHMP2A. (F) Membranous PD-L1 expression in these cells was detected by flow cytometry. All results are expressed as mean ± SD of 3 independent experiments. *P < .05 compared with empty vector; #P < .05 compared with STAT3WT.
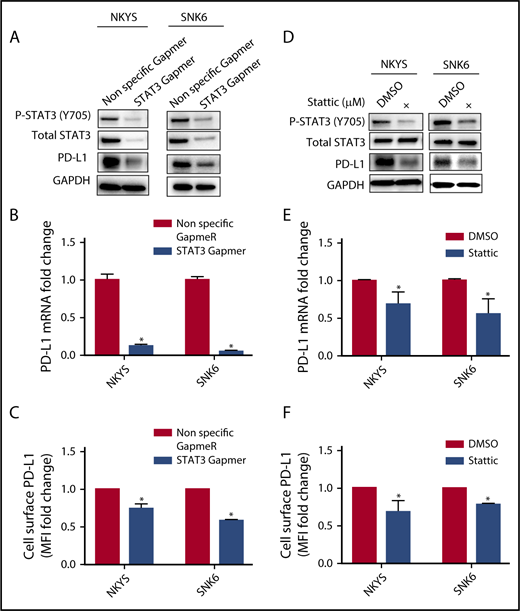
Effect of STAT3 silencing and inhibition on PD-L1 expression in STAT3 mutant NKTL cell lines. STAT3 mutant NKTL cell lines NKYS and SNK6 cells were nucleofected with nonspecific and STAT3-specific Gapmer for 72 hours. These cells were harvested for (A) western blot analysis of pSTAT3, total STAT3, and PD-L1; (B) RT-qPCR to detect PD-L1 mRNA; and (C) flow cytometry to detect membranous PD-L1 expression. NKYS and SNK6 were incubated with dimethyl sulfoxide vehicle or 1 µM Stattic for 24 hours. These cells were harvested for (D) western blot for pSTAT3, total STAT3, and PD-L1; (E) RT-qPCR to detect PD-L1 mRNA; and (F) flow cytometry to detect membranous PD-L1 expression. PD-L1 mRNA was represented as fold change relative to control and normalized against housekeeping gene CHMP2A. Membranous PD-L1 was represented as MFI fold change relative to control. All results are expressed as mean ± SD of 3 independent experiments. *P < .05 compared with control.
Effect of STAT3 silencing and inhibition on PD-L1 expression in STAT3 mutant NKTL cell lines. STAT3 mutant NKTL cell lines NKYS and SNK6 cells were nucleofected with nonspecific and STAT3-specific Gapmer for 72 hours. These cells were harvested for (A) western blot analysis of pSTAT3, total STAT3, and PD-L1; (B) RT-qPCR to detect PD-L1 mRNA; and (C) flow cytometry to detect membranous PD-L1 expression. NKYS and SNK6 were incubated with dimethyl sulfoxide vehicle or 1 µM Stattic for 24 hours. These cells were harvested for (D) western blot for pSTAT3, total STAT3, and PD-L1; (E) RT-qPCR to detect PD-L1 mRNA; and (F) flow cytometry to detect membranous PD-L1 expression. PD-L1 mRNA was represented as fold change relative to control and normalized against housekeeping gene CHMP2A. Membranous PD-L1 was represented as MFI fold change relative to control. All results are expressed as mean ± SD of 3 independent experiments. *P < .05 compared with control.
To understand the underlying mechanism of STAT3 regulation of PD-L1 expression, chromatin immunoprecipitation qPCR assay was performed using STAT3 antibody in Ba/F3 cells expressing p.E616K. We observed a robust increase in occupancy in p.E616K mutant compared with STAT3WT and empty vector (supplemental Figure 6). This suggests that stronger STAT3 binding resulting from the mutation may increase the transcriptional activity of PD-L1 gene promoter and lead to higher expression of PD-L1 in p.E616K mutant.
STAT3 activation correlates significantly to PD-L1 expression in NKTL tumors
To determine the clinical relevance of STAT3 activation and PD-L1 expression, we performed immunohistochemistry staining in 30 NKTL tumors with STAT3WT and STAT3 mutations, including novel (p.E616G, p.E616K, and p.E696K) and hotspot variants. In 9 STAT3 mutant patients, the nuclei of malignant cells were positively stained with pSTAT3 antibody in all cases, supporting in vitro results that novel STAT3 mutants were constitutively phosphorylated and translocated to the nucleus (Figure 5 and data not shown). Interestingly, STAT3 was constitutively phosphorylated in 67% of STAT3WT patients (6/9), suggesting other mechanisms leading to STAT3 dysregulation in the absence of STAT3 mutations. Remarkably, 93% of the NKTL tumors (28/30) were positive for PD-L1 expression, although pSTAT3 was positive in only 77% of tumors (23/30), suggesting the presence of other mechanisms involved in PD-L1 regulation. Expression of PD-L1 was significantly higher in tumors positive for pSTAT3 than in those that were negative, but was not associated with STAT3 mutation status or the clinical characteristics shown in supplemental Figure 7. These results suggested that STAT3 activation is 1 of the contributing factors to high PD-L1 expression in NKTL.
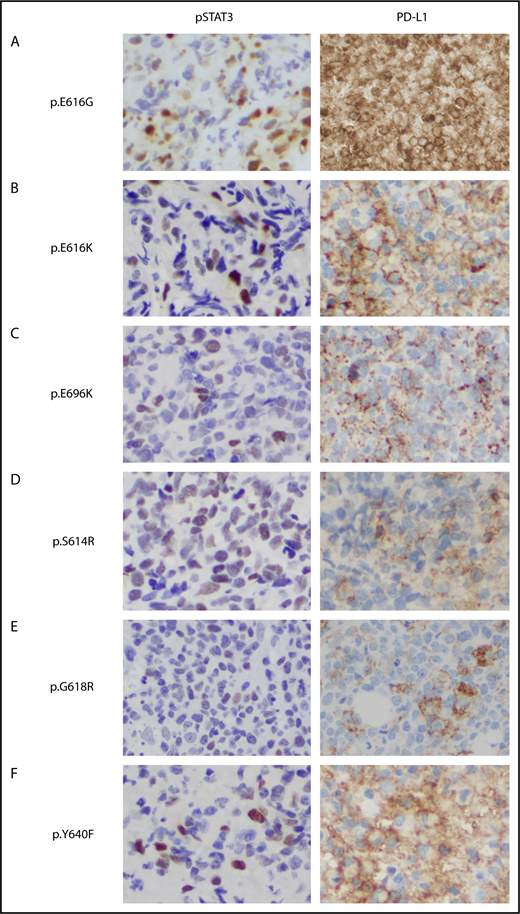
Immunohistochemical staining of phosphorylated STAT3 and PD-L1 in NKTL tumor samples with STAT3 mutations. (A-F) Formalin-fixed, paraffin-embedded sections from tumor biopsy samples of patients with NKTL with novel (p.E616G, p.E616K, p.E696K) and hotspot (p.S614R, p. G618R, p.Y640F) STAT3 mutations that were stained with antibodies against pSTAT3 (left) and membranous PD-L1 (right). Immunohistochemistry shows constitutive phosphorylation and nuclear localization of STAT3 (Y705) associated with high PD-L1 expression in the STAT3 mutant cases (brown staining). Original magnification, ×400.
Immunohistochemical staining of phosphorylated STAT3 and PD-L1 in NKTL tumor samples with STAT3 mutations. (A-F) Formalin-fixed, paraffin-embedded sections from tumor biopsy samples of patients with NKTL with novel (p.E616G, p.E616K, p.E696K) and hotspot (p.S614R, p. G618R, p.Y640F) STAT3 mutations that were stained with antibodies against pSTAT3 (left) and membranous PD-L1 (right). Immunohistochemistry shows constitutive phosphorylation and nuclear localization of STAT3 (Y705) associated with high PD-L1 expression in the STAT3 mutant cases (brown staining). Original magnification, ×400.
Discussion
By means of targeted sequencing, we have demonstrated that alterations in the JAK/STAT signaling pathway are highly prevalent in PTCL and NKTL, especially NKTL (78%), suggesting that targeting the JAK/STAT pathway might be of therapeutic benefit to patients with NKTL and PTCL. High frequencies of JAK/STAT mutations in NKTL have been previously reported, confirming the results of our study (62% [21/34] in Lee et al, 64% [16/25] in Jiang et al, and 48% [12/25] in Dobashi et al).40-42 However, we observed much higher mutation incidence in PTCL-NOS, ALCL, and CTCL subtypes in our cohort when compared with other cohorts,6,43-49 which is most likely explained by the high sequencing depth of our targeted sequencing approach that allowed more sensitive detection of mutations.
The high prevalence of STAT3 mutations in NKTL in our study (21%, 23/109) was similar to the cohort by Lee et al (9/34, 27%),40 but almost twice that of other cohorts (Jiang et el [11%, 11/105], Kucuk et al [12%, 3/25], and Dobashi et al [8%, 2/25]).41,42,50 The TP53 pathway is frequently dysregulated in a variety of neoplasms; however, TP53 mutations are rare (<10%) in PTCL.51-54 In our study, we observed much higher frequency of TP53 aberrations in PTCL-NOS (27%) that coexist with another member in the JAK/STAT pathway, suggesting the interplay between TP53 and JAK/STAT signaling. Indeed, studies have demonstrated that TP53 abnormalities act in collaboration with JAK mutations to drive the transformation of myeloproliferative neoplasms into leukemia55,56 and the role of STAT5 signaling in regulating TP53 expression by MDM2.57
In our study, SOCS-1 missense and nonsense mutations were observed in 6 cases consisting of 4 NKTL and 2 PTCL-NOS. To our knowledge, SOCS-1 mutations have not been reported in PTCL and NKTL, with the exception of a study that found SOCS-1 mutation in 3% (1/34) of primary intestinal T-cell lymphoma cases.7 Mutations in SOCS-1 have largely been implicated in B-cell neoplasms58 and cHL33 as a result of the B-cell-specific aberrant somatic hypermutation process, and were associated with the stabilization of pJAK2 in primary mediastinal B-cell lymphoma59 and accumulation of pSTAT5 in cHL33 However, recent studies have reported the ability of a SOCS-1 to inhibit STAT3 signaling in ALK− ALCL60 and JAK3/STAT5 signaling in CTCL,61 suggesting that SOCS-1 may be a potential target for antitumor therapy in PTCL and NKTL.
As discussed, STAT3 is the most frequently mutated gene in NKTL. We characterized novel STAT3 mutations (p.427H, p.E616G, p.E616K, p.E696K) and provided strong in vitro and in vivo data indicating that they are constitutively active. STAT3 mutants exhibited 2- to 6-fold greater sensitivity to Stattic compared with STAT3WT, suggesting that STAT3 inhibition might be clinically relevant for treating STAT3 mutant NKTL. To determine whether STAT3 mutations were driver mutations that affect cell proliferation and survival, we used an IL-3-dependent murine lymphoid Ba/F3 model that can be transformed to IL-3 independence in the presence of an oncogenic event. Surprisingly, all mutants except p.E616K failed to confer IL-3-independent growth to Ba/F3 cells despite inducing constitutive phosphorylation of STAT3. This phenomenon was also observed in Ba/F3 cells expressing hotspot STAT3 mutation p.Y640F,45,62 shown by previous studies to incur a growth advantage to transduced NKTL cell lines50 and increased STAT3 transcriptional activity in HEK 293 human embryonic kidney cells and HEP3B human hepatoma cells expressing a STAT3-driven luciferase reporter gene.28,38,63 In a recent study, 24 genes identified in the Ba/F3 model as nonfunctional were confirmed as activating in the MCF10A human breast epithelial cell line model, which indicates there may be detection of potential false negatives in the Ba/F3 model.64 Further studies for novel STAT3 mutations in relevant human cell lines and xenograft models are thus warranted to confirm their oncogenicity in NKTL.
We demonstrated that STAT3 is frequently dysregulated in NKTL (77%), consistent with previous studies,65,66 and this was associated with STAT3 mutations in 50% (9/18) of tumor samples. Indeed, studies have reported mechanisms underlying the constitutive activation of STAT3 independent of STAT3 mutations. Gain-of-function mutations involving JAKs have been implicated in activating STAT3 and contributing to the pathogenesis of hematologic malignancies and solid tumors.8,67,68 Cytokines such as IL-6, IL-10, and IL-11 released into the tumor microenvironment by tumor cells have been shown to activate STAT3, which in turn upregulates chemokines, attracting immune and inflammatory cells that further propagate STAT3 activity.69,70 Latent membrane protein 1 (LMP1), an Epstein-Barr virus oncoprotein expressed in NKTL has been reported to mediate activation of STAT3 through its carboxyl-terminal activation domain.71 However in our study, we failed to observe an association between LMP1 expression and STAT3 activation (supplemental Figure 8A-B) in NKTL tumor and cell lines.
To our knowledge, this is the first study to reveal a strong association between high PD-L1 expression and activation of STAT3 pathway in NKTL tumor and cell lines, which is consistent with studies in ALK+ and ALK− ALCL,18,20 multipotent human mesenchymal stromal cells,72 nonsmall cell lung cancers,73 and head and neck squamous cell carcinoma.74 Up to our expectation, PD-L1 was not associated with STAT3 mutation status per se, as STAT3 activation is not driven by STAT3 mutations alone. This suggests a potential synergistic effect when combining STAT3 inhibitors and anti-PD1/PD-L1 antibodies in the treatment of NKTL. Targeting STAT3 has the potential to not only directly inhibit tumor growth but also overcome tumor-induced immunosuppression to enhance antitumor efficacy.
In our study, PD-L1 was positive in 93% of NKTL cases, which is consistent with the study of Jo et al,75 who reported PD-L1 expression of 90%. Other studies reported lower rates (Kim et al, 56%; Han et al, 60%),76,77 which could be attributed to differences in criteria of determining PD-L1 positivity between studies. Together with previous studies, we demonstrated that PD-L1 is aberrantly expressed in NKTL tumors, suggesting that the PD-L1/PD-1 axis may serve as a potential candidate for immunotherapy in NKTL. Further studies are needed to determine whether members of the JAK/STAT pathway other than STAT3 are involved in the regulation of PD-L1 in NKTL. Loss-of-function alterations in JAK1/2 can lead to acquired resistance to anti-PD-1 therapy in patients with melanoma as the result of a lack of adaptive PD-L1 expression from the loss of response to interferon γ signaling.78 Therefore, somatic alterations resulting in activation/deactivation of other JAK members or negative regulators such as SOCS-1 may serve as a pathway for upregulating PD-L1 expression in NKTL and predicting response to PD-1/PD-L1 blockade.
Conclusion
In summary, STAT3 and TP53 were the most frequently mutated genes in the JAK/STAT signaling pathway in PTCL and NKTL, where the incidence of STAT3 and TP53 mutations were highest in NKTL (21%) and PTCL-NOS (27%), respectively. We identified novel activating STAT3 mutations (p.D427H, p.E616K, pE616K, and p.E696K) that might be promising targets for inhibition in the treatment of NKTL. On the basis of our results in cell lines and primary tumor samples, we demonstrated for the first time that STAT3 regulates PD-L1 expression in NKTL, thus providing a rationale for combinations of targeted therapies and immune checkpoint blockade inhibitors in NKTL, and possibly PTCL.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank all the participants in the study, Jeslin Chian Hung Ha, Suk Teng Chin, Rebecca Kee, and Khoo Lay Poh from the Division of Medical Oncology, National Cancer Centre Singapore, for their great assistance in collecting and collating samples and clinical data from patients; staff members at the biobank of Singapore General Hospital, National University Cancer Institute of Singapore, Guangdong Hospital Sun Yat-Sen University Cancer Center for their generous contribution in preparing patient samples; and the Division of Pathology, Singapore General Hospital, SingHealth Flow Cytometry unit, and the SingHealth Tissue Repository for assistance with this project. We also thank Patrick Tan, Xing Manjie, and Xu Chang from Cancer & Stem Cell Biology, Duke-NUS Medical School for their assistance in the chromatin immunoprecipitation qPCR experiment.
This study was supported by research funding from the Singapore Ministry of Health’s National Medical Research Council, Tanoto Foundation as Professorship in Medical Oncology, New Century Foundation Limited, Ling Foundation, and Singapore National Cancer Centre Research Fund, Oncology Academic Clinical Program Cancer Collaborative Scheme, Lee Kong Chian School of Medicine, Nanyang Technological University Singapore Start-Up Grant, and Biomedical Research Council, A*STAR.
Authorship
Contribution: T.L.S., M.-L.N., B.-T.T., S.-T.L., and C.-K.O. provided conception and design; T.L.S., M.-L.N., J.T., Y.L., Z.-M.L., W.-L.P., A.K., G.-C.W., N.K.V., and E.K.-Y.W. provided development of methodology and execution of experiments; T.L.S., J.-Q.L., B.K.-H.C., S.N., N.-F.G., H.F., and C.-K.O. provided analysis and interpretation of data; T.L.S., J.-Q.L., D.-C.H., H.F., S.-T.L., and C.-K.O. provided writing, review, and revision of manuscript; S.-T.L. and C.-K.O. provided study supervision; Y.-H.L., F.Z., H.-L.R., T.T., J.-X.B., S.-B.N., and W.-J.C. provided samples; B.K.-H.C. and D.C. provided material and technical support; and J.I., S.-B.N., and S.-Y.T., supplied pathologic diagnosis
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Choon-Kiat Ong, Lymphoma Genomic Translational Research Laboratory, Division of Medical Oncology, National Cancer Centre Singapore, 11 Hospital Dr, 169610 Singapore, Singapore; e-mail: cmrock@nccs.com.sg; and Soon-Thye Lim, Division of Medical Oncology, National Cancer Centre Singapore, 11 Hospital Dr, 169610 Singapore, Singapore; e-mail: lim.soon.thye@singhealth.com.sg.
