

Key Points
IUHCT induces DST in murine models of sickle cell and β-Thal.
IUHCT combined with postnatal nonmyeloablative allogeneic BM transplants corrects the disease phenotype in SCD and Thal mice.
Abstract
Sickle cell disease (SCD) and thalassemias (Thal) are common congenital disorders, which can be diagnosed early in gestation and result in significant morbidity and mortality. Hematopoietic stem cell transplantation, the only curative therapy for SCD and Thal, is limited by the absence of matched donors and treatment-related toxicities. In utero hematopoietic stem cell transplantation (IUHCT) is a novel nonmyeloablative transplant approach that takes advantage of the immunologic immaturity and normal developmental properties of the fetus to achieve mixed allogeneic chimerism and donor-specific tolerance (DST). We hypothesized that a combined strategy of IUHCT to induce DST, followed by postnatal nonmyeloablative same donor “booster” bone marrow (BM) transplants in murine models of SCD and Thal would result in high levels of allogeneic engraftment and donor hemoglobin (Hb) expression with subsequent phenotypic correction of SCD and Thal. Our results show that: (1) IUHCT is associated with DST and low levels of allogeneic engraftment in the murine SCD and Thal models; (2) low-level chimerism following IUHCT can be enhanced to high-level chimerism and near complete Hb replacement with normal donor Hb with this postnatal “boosting” strategy; and (3) high-level chimerism following IUHCT and postnatal “boosting” results in phenotypic correction in the murine Thal and SCD models. This study supports the potential of IUHCT, combined with a postnatal nonmyelablative “boosting” strategy, to cure Thal and SCD without the toxic conditioning currently required for postnatal transplant regimens while expanding the eligible transplant patient population due to the lack of a restricted donor pool.
Introduction
Sickle cell disease (SCD) and β-thalassemia (Thal) are the two most common hemoglobinopathies worldwide. They are autosomal recessive disorders, which can be diagnosed prenatally. SCD results from a missense mutation in the β-globin gene that causes hemoglobin (Hb) to polymerize resulting in RBC deformation, “sickling,” and subsequent severe hemolytic anemia and vaso-occlusive disease. Thal is caused by genetic defects that reduce β-globin protein levels resulting in severe anemia. Although there have been significant improvements in the management of SCD and Thal, hematopoietic stem cell transplantation (HSCT) remains the only curative treatment.1-3 The standard protocol, a myeloablative HLA-identical matched transplant, is limited by the lack of an HLA-matched donor for most patients as well as graft-versus-host disease (GVHD) and treatment toxicities.4-9 Recent attempts to expand the donor pool by using haploidentical donors and decrease transplant-related toxicities by using minimally myeloablative regimens, although promising, still require immunosuppression and are subject to conditioning-related toxicities.3,4,10-19 SCD and Thal can be phenotypically corrected with lymphomyeloid chimerism levels of ∼20%, highlighting the therapeutic potential of nonmyeloablative regimens.4,20,21
In utero hematopoietic stem cell transplantation (IUHCT) is a nonmyeloablative approach that does not require immunosuppression. Experimentally, IUHCT has been shown to achieve allogeneic mixed hematopoietic chimerism in multiple animal models.22-30 Engraftment is primarily limited by host cell competition, and, in general, IUHCT has resulted in chimerism levels below those anticipated to be therapeutic for most target diseases.31,32 We have previously proposed and validated in the nondiseased murine and canine models, a strategy of IUHCT for the induction of donor-specific tolerance (DST), followed by nonmyeloablative postnatal HSCT to “boost” levels of engraftment into the therapeutic range.29,33,34
Using IUHCT alone, we have previously demonstrated low level mixed hematopoietic chimerism in murine models of hemoglobinopathy.35 Although some improvement was observed, chimerism levels were inadequate to correct the disease phenotype. Because SCD and Thal are likely target disorders for the clinical application of IUHCT, it is important to demonstrate correction of phenotype by a clinically applicable IUHCT-based strategy. In this study, we demonstrate complete correction of the disease phenotype in murine models of SCD and Thal by the two-step strategy of DST induction by IUHCT, followed by same donor, nonmyeloablative postnatal HSCT.
Methods
Animals
SJL/J (H-2Ks, double α-globin), CBA/J (H-2Kk), and C57Bl/6 (H-2Kb, called “B6”) mice were purchased from The Jackson Laboratory. Transgenic mice heterozygous for human α- and β-sickle globins were provided courtesy of Dr Tim Townes and Dr Mohandas Narla.36,37 Upon breeding, heterozygous β-thalassemic mice (Hbbs/Hbbo, H-2Kb, single α-globin, called “Thal mice”), and mouse/human hybrid Hb mice that produce approximately 50% each of human α- and mouse α-globins, and 60% or 100% human βS-globin (H-2Kb, HbβS; 60% or 100% HbβS mice, called “SCD mice”) were generated (see supplemental Table 1 on the Blood Web site). Experimental protocols were approved by The International Animal Care and Use Committee, and followed guidelines set forth in the National Instate of Health Guide for the Care and Use of Laboratory Animals.
Donor bone marrow (BM) harvest and T-cell depletion
BM was harvested from 6- to 8-week-old SJL/J donors as previously described.35 Tibias and femurs were flushed with Ca++/Mg++ free phosphate buffered saline (PBS) (Life Technologies, Grand Island, NY). Single-cell suspensions were filtered through a 70-μm nylon mesh filter and the light density mononuclear cells (LDMCs) were isolated by Ficoll gradient separation (Histopaque-1077; Sigma-Aldrich, St. Louis, MO). The LDMCs were T-cell depleted using anti-CD3 fluorescein isothiocyanate (FITC) antibody (BD Pharmingen, San Diego, CA), anti-FITC microbeads, and a VarioMACS magnetic cell sorter (Miltenyi Biotec, Auburn, CA) to obtain a CD3+ fraction <0.5%. Cells were resuspended in PBS (1 × 106 cells/μL) after ensuring >95% viability by trypan blue exclusion.
IUHCT
Fetuses of timed dated SCD, Thal, or B6 mice were injected on gestational day 14 as previously described.35 Briefly, anesthetized mice underwent a midline laparotomy and the uterine horns were exposed. Fetal intraperitoneal injection of 5 × 106 SJL/J BM cells in 5 μL PBS was performed under direct vision with a 100-μm outside diameter beveled micropipette.
Postnatal BM transplant (BMT)
Peripheral blood (PB) of IUHCT recipients was analyzed by flow cytometry at 4 weeks of age to identify chimeric animals. Chimeric SCD, Thal, and B6 mice received either 0 cGy, 82 cGy, or 138 cGy total body irradiation (TBI) at 4 or 32 weeks of age. Six to 8 hours after TBI, 30 × 106 SJL/J BM cells were injected via tail vein. Controls consisted of age matched Thal or SCD mice that had not undergone IUHCT. TBI was delivered as a single dose by a Gammator M-38 137Cs irradiator at a rate of 276 cGy/min.
Analysis of donor mononuclear cell (MNC) chimerism and multilineage engraftment
Chimerism levels were assessed in recipients of IUHCT at 4 weeks of age and then monthly until postnatal BMT. After postnatal BMT, chimerism levels were assessed every 1 to 2 months until completion of the study. Donor cell lineage analysis was performed at 28 weeks of age in 4-week-old recipients of a postnatal BMT. For all analyses, ∼200 μl of PB was collected in heparinized capillary tubes via retro-orbital vein puncture and diluted to 10 ml with heparinized PBS. LDMCs were collected and flow cytometry performed on a FACScan (Becton Dickenson, Mountain View, CA). Antibodies used for analysis included anti-H2Ks FITC, anti-H2Kb phycoerythrin, and biotin conjugated anti-CD3, anti-B220, anti-GR1, and anti-CD11b, which were developed with streptavidin-cytochrome (BD Pharmingen). Propidium iodide staining was used to exclude dead cells.
Mixed lymphocyte reaction (MLR)
Splenocyte responder cells were harvested from allogeneic chimeric mice created by IUHCT prior to postnatal BMT; 2 × 105 splenic LDMCs were cultured for 3 days with 5 × 105 stimulators (treated with mitomycin C) at 37°C in 5% CO2 in RPMI 1640 medium (Life Technologies) supplemented with 10% fetal bovine serum (Life Technologies), 50 mM 2-mercaptoethanol (Sigma), and antibiotics (penicillin, 100 U/mL and streptomycin, 100 mg/mL) (Life Technologies). Experiments were performed in triplicate. Stimulators consisted of irradiated splenocytes from either B6, SJL/J, or third-party CBA mice. After culture, cells were pulsed with 3H-thymidine and collected 24 hours later. Stimulation indices were calculated by dividing mean cpm by mean background cpm (with no stimulator cell population).
GVHD assessment
Recipients of postnatal BMT were monitored for GVHD manifested by weight loss, alopecia, ragged fur, hunched appearance, diarrhea, and/or decreased activity. Histologic examination following hematoxylin and eosin staining of skin, liver, intestine, and spleen was performed in chimeric mice 24 weeks after postnatal BMT.
Donor Hb expression
Donor Hb expression was assessed by reverse-phase high-performance liquid chromatography (HPLC). Evaluation of donor Hb was initially performed at 4 weeks and then monthly until 24 to 56 weeks of age. PB was centrifuged and RBCs were washed twice in saline; 20 μl aliquot of cells was lysed in 100 μl of hemolyzin solution for 10 minutes. After the addition of 1:10 volume of 10% NaCl solution, ghosts were removed by centrifugation (15 000 g × 15 minutes). HPLC was performed by injecting Hb samples on a Hitachi D-7000 HSM Series apparatus (Hitachi Instruments, San Jose, CA) using a Jupiter 5 m C-4 250 × 4.6 mm column (Phenomenex, Torrence, CA) and a gradient from 20% to 60% acetonitrile in 0.3% trifluoroacetic acid in 35 minutes, with UV detection at 215 nm. Hb of the donor mouse strain has double α-globin chains with 0.6:1 ratio (α1-globin: α2-globin). The percent donor Hb was calculated using the peak representing α2-globin (supplemental Figure 1):
- (1) Chimeric SCD mouse:
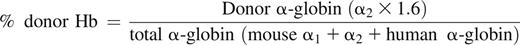
- (2) Chimeric Thal mouse:

Immunofluorescence staining for ΗbβS
Immunofluorescence staining using the anti-human HbβS antibody was performed as previously described to determine the HbβS level in chimeric mice with greater accuracy.38,39 Monolayer smears of RBCs with uniform thickness were prepared using a DiffSpin (StatSpin Technologies, Norwood, MA) slide maker to eliminate factors affecting fluorescence intensity from individual cells. RBCs were washed by centrifugation with a buffer (125 mM NaCl, 5 mM KCl, 5 mM MgCl2, and 10 mM phosphate, pH 7.4 at room temperature), and then fetal bovine serum was added (2:1 ratio). Prepared smears were fixed with an acetone, methanol, and ethanol (3:1:1) solution and washed in PBS followed by distilled water. An anti-human HbβS monoclonal antibody (PerkinElmer Wallac Inc., Norton, OH) was applied, and smears were incubated in a humidified chamber (30 minutes at 37°C) and subsequently washed in PBS followed by distilled water. Superimposition of the fluorescent image with that containing all cells in the same field via a bandpass filter (415 ± 15 nm; Omega Optical, Inc., Brattleboro, VT) allowed the determination of the percent HbβS staining (ARGUS50; Hamamatsu, Bridgewater, NJ).
Quantification of iron deposition in spleen
Naïve Thal, B6, and Thal chimeric mice were euthanized at 40 weeks of age and splenic iron deposition was assessed as a measure of hemolytic or vaso-occlusive disease. Spleens were fixed in 10% buffered formalin and embedded in paraffin. Sections were stained with Perls’ iron stain method. Deparaffinized and hydrated slides were placed in mixed 20% hydrochloric acid and 10% potassium ferrocyanide stock solution (1:1) for 30 minutes. Slides were rinsed and then counterstained with nuclear fast red solution and examined by light microscopy.
Urine concentrating ability
Urine concentrating ability was measured after water deprivation for 24 hours. Mice were placed on parafilm, and urine samples collected and diluted 10-fold. A 20 μl aliquot was analyzed with a 3300 Micro-Osmometer (Advanced Instruments, Inc., Norwood, MA). Samples were analyzed in triplicate.
Functional and morphologic analysis of RBCs
Functional and morphologic analyses of RBCs in control and chimeric mice are summarized in supplemental Methods.
Statistical analysis
Data are represented as the mean ± 1 SD unless otherwise indicated. Statistical comparisons between groups were performed with the Student t test for 2 samples with unequal variances. A 2-tailed P ≤ .05 was considered significant. Fisher’s exact test was used to compare factors expressed as categorical values.
Results
IUHCT alone results in long-term multilineage allogeneic engraftment associated with high donor Hb expression and DST in SCD and Thal mice
Survival to weaning following IUHCT was ∼60% and 20% in nonhemoglobinopathy and hemoglobinopathy mice, respectively. Forty-nine percent of all mice had chimerism levels >1% at the initial 1-month time point (38% of wild-type [WT] vs 54% of hemoglobinopathy mice; P = .13). Consistent with our previous study,35 allogeneic engraftment after IUHCT alone was stable at 40 weeks posttransplant in SCD, Thal, and B6 recipients (Figure 1A). Chimerism was multilineage at 28 weeks of age consistent with engraftment of HSCs (Figure 1C). The SCD and Thal environment provided a competitive advantage for donor RBCs. Donor Hb levels following IUHCT are higher than the corresponding level of MNC engraftment in SCD and Thal mice but not B6 mice (Figure 1B vs 1A), with Hb levels significantly higher in SCD and Thal recipients compared with B6 recipients (Figure 1B).

Donor-cell engraftment, Hb levels, and DST following IUHCT. (A) PB of SCD, Thal, and B6 WT recipients of an IUHCT was analyzed for % total donor MNC engraftment and (B) % donor Hb. (A) *P < .05 for SCD vs WT; (B) Hb levels were statistically different with P < .05 between SCD vs WT and Thal vs WT with the exception of those marked by #; error bars: SEM. (C) At 6 months of life, PB was assessed for donor lymphomyeloid engraftment. (D) DST of chimeric mice following IUHCT was assessed by in vitro MLR. Responder splenocytes from chimeric SCD, chimeric Thal, and naïve B6 mice were assessed for proliferation against B6 (self), SJL/J (donor), and CBA (third-party) stimulators.
Donor-cell engraftment, Hb levels, and DST following IUHCT. (A) PB of SCD, Thal, and B6 WT recipients of an IUHCT was analyzed for % total donor MNC engraftment and (B) % donor Hb. (A) *P < .05 for SCD vs WT; (B) Hb levels were statistically different with P < .05 between SCD vs WT and Thal vs WT with the exception of those marked by #; error bars: SEM. (C) At 6 months of life, PB was assessed for donor lymphomyeloid engraftment. (D) DST of chimeric mice following IUHCT was assessed by in vitro MLR. Responder splenocytes from chimeric SCD, chimeric Thal, and naïve B6 mice were assessed for proliferation against B6 (self), SJL/J (donor), and CBA (third-party) stimulators.
DST is required for enhancement of chimerism by nonmyeloablative postnatal same donor BMT. MLRs were performed using responder splenocytes from chimeric Thal and SCD mice (chimerism level >1%) following IUHCT, as well as B6 mice that had not received an IUHCT. Unlike B6 splenocytes, chimeric Thal and SCD splenocytes did not respond to SJL/J (donor) stimulator cells (Figure 1D). All 3 responder populations proliferated in response to third-party stimulators and demonstrated minimal stimulation to “self.” In general, there is good correlation between a nonreactive MLR and skin graft tolerance in the murine model following IUHCT.40,41 These results confirm that even low levels of engraftment after IUHCT are associated with DST in SCD and Thal mice, and should provide a platform on which nonmyeloablative postnatal transplants can be successfully performed.
Enhancement of engraftment with a nonmyeloablative postnatal transplant following IUHCT
We previously demonstrated the ability to enhance allogeneic engraftment in a normal mouse model by 3 different nonmyeloablative conditioning protocols.33,34,42 In the current study, we used TBI because of its easily titratable dosing even at different ages.34 Engraftment enhancement was successful in 100% of mice with chimerism levels >1% and 76% of mice with chimerism levels <1% in agreement with our previous study.33 Enhancement was TBI dose dependent and similar between the 3 recipient strains (Figure 2A). Enhanced donor-cell engraftment was multilineage (at 28 weeks of age) and stable at 6 to 10 months of life (Figure 2C). Similar to mice receiving a postnatal transplant at 4 weeks of age, chimeric mice transplanted at 32 weeks demonstrated a TBI dose-dependent enhancement of engraftment (Figure 2B). Of note, engraftment enhancement at 4 weeks was higher than at 32 weeks (∼70% vs 40% to 50%; 138 cGy, final time point) likely representing the increased cell dose/kg in the 4-week-old mice. Mice that did not receive an IUHCT failed to demonstrate engraftment following postnatal transplants (data not shown) in agreement with our previous studies in which tolerance achieved though IUHCT was required to enhance engraftment.34

Enhancement of engraftment by a nonmyeloablative postnatal “boosting” regimen in recipients of IUHCT. Chimeric SCD, Thal, and B6 WT mice were boosted at (A) 4 weeks or (B) 32 weeks of age following conditioning with one of 3 doses of irradiation (138 cGy, 82 cGy, and 0 cGy). Engraftment levels of control chimeric SCD, Thal, and WT mice that did not receive a “booster” transplant are included in (B) for comparison. Error bars in (A) and (B) are expressed as SEM. Levels of chimerism within each group (WT, Thal, and SCD) are statistically different (P < .05) following boosting compared with the nonboosted control at that time point, with the exception of the points marked *. Levels of chimerism were statistically different between each irradiation dose within each group (WT, Thal, and SCD) at the indicated time point with the exception of the points marked # (WT, 82 cGy vs 138 cGy), $ (Thal, 82 cGy vs 138 cGy), and & (SCD, 4 weeks, 0 cGy vs 82 cGy; 32 weeks, 82 cGy vs 138 cGy). (C) PB was assessed for multilineage donor-cell engraftment at 28 weeks of age in naïve SJL/J donor mice and chimeric SCD, Thal, and WT mice following postnatal “booster” transplants with 138 cGy conditioning at 4 weeks of age.
Enhancement of engraftment by a nonmyeloablative postnatal “boosting” regimen in recipients of IUHCT. Chimeric SCD, Thal, and B6 WT mice were boosted at (A) 4 weeks or (B) 32 weeks of age following conditioning with one of 3 doses of irradiation (138 cGy, 82 cGy, and 0 cGy). Engraftment levels of control chimeric SCD, Thal, and WT mice that did not receive a “booster” transplant are included in (B) for comparison. Error bars in (A) and (B) are expressed as SEM. Levels of chimerism within each group (WT, Thal, and SCD) are statistically different (P < .05) following boosting compared with the nonboosted control at that time point, with the exception of the points marked *. Levels of chimerism were statistically different between each irradiation dose within each group (WT, Thal, and SCD) at the indicated time point with the exception of the points marked # (WT, 82 cGy vs 138 cGy), $ (Thal, 82 cGy vs 138 cGy), and & (SCD, 4 weeks, 0 cGy vs 82 cGy; 32 weeks, 82 cGy vs 138 cGy). (C) PB was assessed for multilineage donor-cell engraftment at 28 weeks of age in naïve SJL/J donor mice and chimeric SCD, Thal, and WT mice following postnatal “booster” transplants with 138 cGy conditioning at 4 weeks of age.
No evidence of GVHD in high chimeric hemoglobinopathy mice
No animal demonstrated evidence of GVHD. Animals in all groups demonstrated stable or appropriate weight gain following postnatal transplants (supplemental Figure 2). Histology on representative animals from each group confirmed no evidence of GVHD (data not shown).
Enhanced donor Hb expression following postnatal transplants in recipients of IUHCT
Correction of hemoglobinopathy phenotypes requires high levels of normal Hb. Chimeric mice demonstrated a dramatic increase in donor Hb expression following postnatal transplants (Figure 3). In those transplanted at 4 weeks, donor Hb constituted >90% of all Hb in SCD, Thal, and B6 recipients following conditioning with either 82 cGy or 138 cGy. This represents an increase over total donor MNC engraftment under these conditioning regimens (Figure 3B vs Figure 2A). Although a survival advantage for donor RBCs is expected in the hemoglobinopathy models, the increased erythroid compared with lymphomyeloid engraftment in B6 mice was unexpected and may be strain specific. Of note, an increase in donor Hb expression to clinically relevant levels was also noted in recipients of unconditioned postnatal transplants. The increase in Hb was higher than that of lymphomyeloid engraftment suggesting that an even less intense conditioning regimen may be adequate. Levels of donor Hb expression in mice boosted at 32 weeks were also higher than the lymphomyeloid engraftment at corresponding doses of TBI (Figure 3C vs Figure 2C). The enhanced expression of donor Hb was stable and persisted for 24 to 32 weeks after postnatal transplants in mice transplanted at 32 and 4 weeks of age, respectively.

Long-term high expression of donor Hb following nonmyeloablative postnatal “booster” transplants in chimeric hemoglobinopathy mice. (A) Representive HPLC data, with donor Hb represented by the α2-globin level (green, cross-hatched peak), from chimeric SCD (left) and Thal (right) mice after postnatal boosting following 138 cGy TBI. Donor Hb expression in chimeric mice following IUHCT + boost at (B) 4 or (C) 32 weeks of age was assessed by HPLC. Donor Hb levels within each group (WT, Thal, and SCD) are statistically different (P < .05) following boosting compared with the nonboosted control at that time point with the exception of the points marked *. Donor Hb levels were statistically different at all time points following boosting within each group (WT, Thal, and SCD) between 0 cGy vs 138 cGy, and were statistically different when comparing levels at two sequential irradiation doses (ie, 0 cGy vs 82 cGy or 82 cGy vs 138 cGy) with the exception of those marked # (WT), $ (Thal), and & (SCD). Error bars expressed as SEM.
Long-term high expression of donor Hb following nonmyeloablative postnatal “booster” transplants in chimeric hemoglobinopathy mice. (A) Representive HPLC data, with donor Hb represented by the α2-globin level (green, cross-hatched peak), from chimeric SCD (left) and Thal (right) mice after postnatal boosting following 138 cGy TBI. Donor Hb expression in chimeric mice following IUHCT + boost at (B) 4 or (C) 32 weeks of age was assessed by HPLC. Donor Hb levels within each group (WT, Thal, and SCD) are statistically different (P < .05) following boosting compared with the nonboosted control at that time point with the exception of the points marked *. Donor Hb levels were statistically different at all time points following boosting within each group (WT, Thal, and SCD) between 0 cGy vs 138 cGy, and were statistically different when comparing levels at two sequential irradiation doses (ie, 0 cGy vs 82 cGy or 82 cGy vs 138 cGy) with the exception of those marked # (WT), $ (Thal), and & (SCD). Error bars expressed as SEM.
Quantification of HbβS in RBCs of chimeric SCD mice
Human HbβS expression was assessed to determine the prevalence of recipient RBCs in SCD mice following IUHCT and postnatal BMT. As shown in Figure 4A, SCD chimeric mice following IUHCT alone demonstrated decreased levels of anti-human HbβS staining (∼76.1 to ∼97.2% HbβS+) but a predominance of sickle Hb persisted. In contrast, chimeric SCD mice who underwent a postnatal BMT after conditioning with 82 cGy or 138 cGy TBI demonstrated minimal anti-human HbβS staining (138 cGy: ∼2.0 to ∼8.3% HbβS+; 82 cGy: ∼6.3 to ∼15.0% HbβS+ at 24 weeks) confirming near complete replacement of recipient RBCs with donor RBCs. Levels of immunofluorescent staining inversely correlated with levels of donor Hb detected by HPLC (Figure 4B).

Quantification of ΗbβS+ RBCs in SCD recipients of IUHCT with or without a nonmyeloablative postnatal “booster” transplant. RBCs from naïve SCD mice, chimeric SCD mice following IUHCT, chimeric SCD mice that underwent a postnatal “booster” transplant following conditioning with 138 cGy TBI, and naïve SJL/J mice were stained with a FITC conjugated anti-human ΗbβS antibody. Representative immunofluorescence images demonstrate no staining of RBCs from SJL/J mice, whereas all RBCs from naïve SCD mice demonstrate positive staining. (A) Chimeric SCD mice following IUHCT alone or IUHCT + postnatal “booster” transplant demonstrated progressively less ΗbβS antibody staining, respectively. (B) The degree of anti-HbβS antibody staining in chimeric SCD mice inversely correlated with the percentage of donor Hb noted on HPLC analysis.
Quantification of ΗbβS+ RBCs in SCD recipients of IUHCT with or without a nonmyeloablative postnatal “booster” transplant. RBCs from naïve SCD mice, chimeric SCD mice following IUHCT, chimeric SCD mice that underwent a postnatal “booster” transplant following conditioning with 138 cGy TBI, and naïve SJL/J mice were stained with a FITC conjugated anti-human ΗbβS antibody. Representative immunofluorescence images demonstrate no staining of RBCs from SJL/J mice, whereas all RBCs from naïve SCD mice demonstrate positive staining. (A) Chimeric SCD mice following IUHCT alone or IUHCT + postnatal “booster” transplant demonstrated progressively less ΗbβS antibody staining, respectively. (B) The degree of anti-HbβS antibody staining in chimeric SCD mice inversely correlated with the percentage of donor Hb noted on HPLC analysis.
End organ evaluation in hemoglobinopathy mice following IUHCT and postnatal transplants
Spleens from naïve SCD and Thal mice are larger compared with B6 mice. The spleen size from chimeric SCD and Thal mice following IUHCT and a postnatal transplant was significantly smaller than nonchimeric naïve SCD and Thal mice, and approached the size of B6 mice (Figure 5A-B). Histologic evaluation of the spleens of chimeric Thal mice (which had the greatest reduction in size) demonstrated less iron deposition compared with those from naïve Thal mice and were similar in appearance to B6 control mice (Figure 5C).

Splenic weight and iron deposition in chimeric hemoglobinopathy mice following IUHCT + postnatal BMT. Spleens from chimeric SCD (n = 4) and Thal (n = 5) mice who had undergone a postnatal “booster” transplant were harvested at 40 weeks of age and their weight was compared with age-matched naïve SCD mice (n = 6), Thal mice (n = 7), B6 WT (n = 6), and B6 WT chimeric mice boosted following 138 cGy TBI (n = 4). *P < .05 vs corresponding naïve SCD or Thal mice (A-B). Spleens were evaluated by histology for iron deposition in chimeric Thal mice following postnatal BMT and compared with age-matched naïve Thal and chimeric B6 WT controls (C).
Splenic weight and iron deposition in chimeric hemoglobinopathy mice following IUHCT + postnatal BMT. Spleens from chimeric SCD (n = 4) and Thal (n = 5) mice who had undergone a postnatal “booster” transplant were harvested at 40 weeks of age and their weight was compared with age-matched naïve SCD mice (n = 6), Thal mice (n = 7), B6 WT (n = 6), and B6 WT chimeric mice boosted following 138 cGy TBI (n = 4). *P < .05 vs corresponding naïve SCD or Thal mice (A-B). Spleens were evaluated by histology for iron deposition in chimeric Thal mice following postnatal BMT and compared with age-matched naïve Thal and chimeric B6 WT controls (C).
SCD mice, similar to humans with SCD, have defective urine concentrating ability and develop significant nephropathy.43,44 Urine concentrating ability was assessed in naïve SCD, B6, and chimieric SCD mice that did and did not receive a postnatal transplant. There was a significant increase in urine concentrating ability in chimeric SCD mice following a postnatal transplant compared with chimeric SCD mice following IUHCT alone and naïve SCD mice. The urine osmolarity in “boosted” chimeric SCD mice was similar to that of B6 mice (Figure 6).

Urine concentrating ability in chimeric SCD mice following IUHCT ± postnatal BMT. Urine osmolality was determined after 24 hours of water deprivation in naïve SCD mice (n = 5), naïve B6 mice (n = 4), chimeric SCD mice following IUHCT (n = 4), and chimeric SCD mice who underwent a postnatal boost following IUHCT (n = 4). *P < .05 vs naïve SCD mice.
Urine concentrating ability in chimeric SCD mice following IUHCT ± postnatal BMT. Urine osmolality was determined after 24 hours of water deprivation in naïve SCD mice (n = 5), naïve B6 mice (n = 4), chimeric SCD mice following IUHCT (n = 4), and chimeric SCD mice who underwent a postnatal boost following IUHCT (n = 4). *P < .05 vs naïve SCD mice.
Together with studies shown in supplemental data (supplemental Table 2; supplemental Figures 3-7), these splenic and renal studies suggest that the combined approach of allogeneic IUHCT and nonmyeloablative postnatal transplants corrects the phenotype in the murine SCD and Thal models.
Functional and morphologic assessment of RBCs in hemoglobinopathy mice following IUHCT with or without postnatal transplants
Although a trend toward improved functional and morphologic parameters was seen following IUHCT alone, IUHCT and postnatal transplant significantly improved parameters to measurements similar to control non-hemoglobinopathy mice. Specifically, we demonstrate improved Hb levels, increased RBC half-life, improved RBC morphology including decreased sickling cells in SCD mice, increased RBC stability as indicated by decreased membrane associated denatured Hb levels, and increased oxygen affinity (in SCD mice) in hemoglobinopathy mice following IUHCT with postnatal transplants (supplemental Table 2; supplemental Figures 3-7).
Discussion
IUHCT takes advantage of normal developmental ontogeny to engraft allogeneic cells across immune barriers without myeloablation or immunosuppression resulting in mixed chimerism and DST.28,33,34 Therefore, IUHCT may be applicable to any disorder, such as SCD and Thal, that can be prenatally diagnosed and treated by mixed chimerism. We previously demonstrated the ability to obtain stable allogeneic engraftment following IUHCT using donor fetal liver or BM cells in the murine SCD and Thal models.35 Although encouraging, levels of chimerism achieved by IUHCT alone did not correct phenotype. Despite higher levels of donor Hb (13% to 35%) than donor MNC engraftment (1.1% to 4%), a significant number of sickling cells remained. This is in agreement with studies demonstrating a threshold of 70% and 40% donor myeloid engraftment needed to eliminate peripheral RBC sickling and anemia, respectively.45 Thus, it is likely that clinical strategies to obtain higher levels of donor engraftment after IUHCT will be required to achieve a therapeutic effect.
In the current study, we use DST induced by IUHCT to facilitate postnatal enhancement of engraftment by nonmyeloablative BMT and correct the phenotype of murine hemoglobinopathy models. We intentionally generated low-level chimeric mice by IUHCT using the intraperitoneal mode of transplantation. This protocol results in sustained, long-term allogeneic engraftment that is too low to correct the phenotype but above the threshold for DST. We then applied a low-dose TBI regimen, previously shown to be nonmyeloablative,34 to our chimeric animals for the postnatal transplant. This strategy enhanced donor lymphomyeloid engraftment to levels >40% and 70% at the lowest and highest doses of TBI, respectively. Significantly, these engraftment levels were associated with >90% donor Hb and were sufficient to correct the hemoglobinopathy phenotypes experimentally and presumably clinically. Hemoglobinopathy recipients of IUHCT and a nonconditioned postnatal transplant also demonstrated clinically relevant donor Hb levels suggesting that no or minimal postnatal conditioning may be required to achieve a therapeutic effect. Although preferential engraftment of a common myeloid vs lymphoid progenitor may explain the increased erythroid vs total MNC engraftment, the discrepancy is likely related to a survival advantage of donor RBC in the murine hemoglobinopathy models. This is supported by the increased half-life of RBCs from SJL donors and chimeric hemoglobinopathy mice compared with RBCs from naïve hemoglobinopathy mice (supplemental Table 2). Additional studies in hemoglobinopathies demonstrating decreased apoptosis of normal donor erythroid progenitors, longer normal RBC survival, and similar findings of higher donor erythroid engraftment following mixed chimerism in patients with Thal and SCD also support this hypothesis.3,20,46-49 These results are encouraging and support the potential of more clinically applicable nonmyeloablative conditioning regimens, such as low-dose Busulfan, which we have successfully used in the normal murine and canine models of IUHCT and postnatal transplant, as a strategy for clinical treatment of SCD and Thal.29,33
We used the current SCD model because the presence of the murine/human chimeric Hb limits the disease severity and allows for more stable breeding and survival following fetal intervention in this proof-of-principle study. Unlike the β-Thal model, which has a dramatically reduced RBC half-life and phenotypically resembles human β-Thal (similar postnatal microcytic anemia, splenomegaly, and end-organ damage), the SCD mice have a mild phenotype under normal conditions and require hypoxia to exhibit a more severe phenotype.36,50 Thus, we performed a more detailed functional and morphologic RBC analysis to demonstrate phenotypic correction. An additional limitation of both the SCD and Thal models in this study is that they do not exhibit normal human erythropoietic ontogeny in which there is no dependence on β-globin until after birth. Thus, homozygous mice may be anemic, conferring an engraftment advantage following IUHCT not present in the human.51 Although this advantage may allow for higher initial levels of erythroid engraftment, we do not believe that this advantage is necessary to achieve engraftment following IUHCT, as we have previously demonstrated stable macrochimeric levels of allogeneic engraftment in nondisease mouse models in which no selective advantage exists.28 Furthermore, the purpose of the current study is to demonstrate the ability to achieve levels of donor-cell engraftment adequate for phenotypic correction of the hemoglobinopathies by a clinically relevant IUHCT-based strategy. Because the postnatal transplants are performed after the transition to β-globin production, there is no discrepant selective engraftment advantage after birth in the murine model.
The correction of disease phenotype by higher levels of mixed chimerism is consistent with clinical studies demonstrating the amelioration of SCD and Thal with donor-cell engraftment as low as 15% to 20%.6,21,52,53 Currently, the only curative treatment of SCD or Thal patients is a BMT. A matched-donor transplant following myeloablative conditioning remains the gold-standard against which other transplant regimens are judged.4-6,8,21,54 Advances in conditioning protocols have increased the success of matched myeloablative transplants, but limitations still remain. Overall and event-free survival rates in the largest study to date of myeloablative stem cell transplants for SCD were 93% and 86%, respectively. Despite good survival rates, there remained significant treatment-related morbidity (acute GVHD: 20%; chronic GVHD: 12.6%).5 Similarly, large studies in Thal patients treated with matched myeloablative transplants demonstrate probabilities of overall survival, disease-free survival, rejection-related mortality, and transplant-related mortality of 65% to 79%, 62% to 70%, 4% to 30%, and 4% to 37%, respectively.3,55-59 Assumed within these transplant regimens is the ability to find an HLA-matched donor. It is estimated that up 50% to 60% of Thal patients lack a matched donor and only 21% and 6% of SCD patients had a matched related or unrelated donor, respectively.3,12,60 These limitations, combined with the realization that low levels of engraftment are sufficient to ameliorate disease, have ushered in attempts to minimize myeloablative conditioning and use haploidentical donors. Results from matched donor nonmyeloablative approaches are encouraging and demonstrate minimal treatment-related toxicities with varying degrees of sustained engraftment.3,12,18 Similarly, the use of myeloablative haploidentical regimens to treat Thal patients resulted in graft acceptance and cure in 71% of patients but with the expected treatment-related toxicities.61,62 A recent study combining a nonmyeloablative regimen with haploidentical donors resulted in minimal treatment-related toxicity but 43% graft rejection.10
These studies emphasize the need for increased investigation into alternative transplant approaches, such as IUHCT, that will expand the donor pool while avoiding treatment-related toxicities. IUHCT has additional advantages including the small size of the anticipated human recipient (13 to 14 weeks gestation, ∼35 g), which maximizes the cell dose/kg and the potential decreased risk of GVHD secondary to a robust T-regulatory cell response after IUHCT.63 These advantages make the cell dose used in the current proof-of-principle study clinically practical. Specifically, we recently demonstrated in the preclinical canine model that IUHCT using equivalent HSC doses (5.7 × 108 to 1.7 × 109 CD34+ cells/kg; 2.5 × 108 to 4.1 × 108 CD3+ cells/kg) to those used in the current study may achieve levels of engraftment sufficient to treat target diseases without GVHD.30 Alternatively, if insufficient levels are reached, the current study supports the strategy of IUHCT to induce DST to facilitate nonmyeloablative postnatal transplantation as a clinical approach for treatment of the hemoglobinopathies.29,33,34
The online version of this article contains a data supplement.
There is an on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Kazumi Horiuchi for providing technical advice with the fluorescence imaging assays.
This study was supported by grants from the National Institutes of Health, National Heart, Lung, and Blood Institute (R01 HL64715) (A.W.F.), funds from the Robert J. Kahn Foundation, and the Ruth and Tristram C. Colket, Jr Chair of Pediatric Surgery (A.W.F.).
Authorship
Contribution: W.H.P. designed and performed the research, analyzed the data, and wrote the manuscript; S.H. designed and performed the research, provided vital technical skills, and analyzed the data; O.A. designed and conducted the research, analyzed the data, and wrote the manuscript; Q.C. and A.M. performed the research and contributed vital technical skills; T.A. designed the research and contributed vital analytical skills; and A.W.F. designed the research, contributed vital analytical skills, and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Dr Toshio Asakura died on May 21, 2015.
Correspondence: Alan W. Flake, Department of Surgery, Children’s Hospital of Philadelphia, Abramson Research Center, Room 1116B, 3615 Civic Center Blvd, Philadelphia, PA 19104-4318; e-mail: flake@email.chop.edu.
