

Key Points
Intravenous injection of a foamy virus carrying a corrective gene facilitates immune cell development in a canine model of SCID-X1.
Integration site analysis revealed polyclonal reconstitution in all dogs with evidence for clonal dominance in at least 1 time point.
Abstract
Current approaches to hematopoietic stem cell (HSC) gene therapy involve the collection and ex vivo manipulation of HSCs, a process associated with loss of stem cell multipotency and engraftment potential. An alternative approach for correcting blood-related diseases is the direct intravenous administration of viral vectors, so-called in vivo gene therapy. In this study, we evaluated the safety and efficacy of in vivo gene therapy using a foamy virus vector for the correction of canine X-linked severe combined immunodeficiency (SCID-X1). In newborn SCID-X1 dogs, injection of a foamy virus vector expressing the human IL2RG gene resulted in an expansion of lymphocytes expressing the common γ chain and the development of CD3+ T lymphocytes. CD3+ cells expressed CD4 and CD8 coreceptors, underwent antigen receptor gene rearrangement, and demonstrated functional maturity in response to T-cell mitogens. Retroviral integration site analysis in 4 animals revealed a polyclonal pattern of integration in all dogs with evidence for dominant clones. These results demonstrate that a foamy virus vector can be administered with therapeutic benefit in the SCID-X1 dog, a clinically relevant preclinical model for in vivo gene therapy.
Introduction
Severe combined immunodeficiency (SCID) designates several genetic diseases characterized by a block in T-lymphocyte development, with various forms affecting the development of natural killer cells and/or development and function of B lymphocytes.1 SCID typically presents prior to 6 months of age with severe lymphopenia, recurrent opportunistic infections, and failure to thrive.2,3 Approximately half of all SCID cases are inherited in an X-linked fashion (SCID-X1), secondary to mutation of the common γ chain (γC), a component of the interleukin (IL)-2, IL-4, IL-7, IL-9, IL-15, and IL-21 receptor complexes.4 Due to the multiple defects in cytokine signaling, SCID-X1 is a complex disease that encompasses deficiencies in both cellular and humoral immunity.5 Hematopoietic stem cell (HSC) transplantation is the most common treatment of SCID-X1, but poor stem cell engraftment, morbidity, and mortality remain significant problems in patients receiving HLA-nonidentical transplants.6
Pioneering gene therapy clinical trials for SCID-X1 occurred in the 1990s, fueled in part by the lack of suitable donors for HSC transplantation and from reports of clonal lymphocyte expansion and disease attenuation stemming from natural reversions that result in the expression of functional receptor.7,8 Initial trials used replication-defective murine leukemia virus (MLV)-derived gammaretroviral vectors to deliver the IL2RG gene ex vivo to autologous CD34+ cells in the absence of a conditioning regimen.9,10 This approach was highly effective, resulting in the sustained correction of the disease in the majority of patients enrolled in clinical trials in France and in the United Kingdom.11,12 However, 5 of 20 boys that received gene therapy with the gammaretroviral vector developed clonal T-cell proliferation resulting in leukemogenesis that could be attributed to proviral transactivation of proto-oncogenes, notably LMO2.13,14 These reports emphasized the need for “safer” vector systems that lack the strong enhancer activity of the MLV long terminal repeat and are the primary impetus for the development of self-inactivated lentiviral vectors used in ongoing clinical trials for SCID-X1 patients.15
Foamy viruses comprise a major subfamily of retroviruses with properties that make them attractive for HSC gene therapy. Importantly, foamy viruses are less likely to integrate near proto-oncogenes as compared with MLV-derived gammaretroviral vector systems, and unlike HIV-derived lentiviral vectors do not show an overall preference for insertion within genes.16,17 Foamy viral vectors have exceptionally broad tissue tropism, attributable to the foamy viral envelope receptor, heparan sulfate, expressed ubiquitously on vertebrate cell surfaces.18 This allows them to efficiently transduce human HSC as well as those of nearly all clinically relevant model organisms with an efficiency comparable to standard lentivirus vectors.19 Additionally, foamy viral vectors transduce quiescent cells with higher efficiency than MLV-based vectors.20 These properties contributed to the successful use of foamy viruses for gene replacement therapy in two canine models of human disease: leukocyte adhesion deficiency21,22 and pyruvate kinase deficiency.23 Finally, unlike the vesicular stomatitis virus glycoprotein that is commonly used in lentiviral packaging, the foamy virus envelope resists inactivation by human serum, making it a suitable choice for in vivo gene therapy.24
The development of a safe and effective retroviral vector that can be administered by intravenous injection is an important goal, because it offers a portable and broadly scalable approach for the correction of genetic diseases. Vascular delivery of gammaretroviral vectors has been previously performed in canine models of hemophilia B,25 mucopolysaccharidosis,26 and SCID-X1.27 Furthermore, it has been proposed that in vivo gene therapy for SCID-X1 presents an excellent opportunity to explore the relative safety profiles of different viruses for intravenous delivery.28 Here, we evaluate the efficacy and safety of an intravenous injection of a self-inactivated foamy viral vector for the correction of canine SCID-X1.
Materials and methods
SCID-X1 dogs
All experiments were performed in accordance with protocols approved by the University of Pennsylvania Institutional Animal Care and Use Committees. Details on dogs and breeding strategies are listed in supplemental Methods, available on the Blood Web site.
Generation of the foamy virus vector
The foamy vector pFV.Ef1α.GFP.2A.γC contains enhanced green fluorescent protein (GFP) and the coding sequence for the human γC linked by a T2A element, expressed under control of the elongation factor 1 promoter (Ef1α) (Figure 1A). Foamy virus preparations were produced by polyethylenimine transfection of human embryonic kidney 293 cells as previously described.29 Vector details can be found in supplemental Methods.

Expansion of gene-modified lymphocytes after in vivo injection of a foamy virus vector. (A) Schematic of the foamy viral vector used in the study. EF1α promoter drives the expression of GFP (to evaluate gene marking) and the coding region of the human common γ chain separated by a 2A self-cleaving peptide. Percentage of lymphocytes expressing (B) γC+ and (C) the GFP transgene in 5 study dogs that received foamy virus in vivo gene therapy, plotted as days postinjection. Lymphocytes in panels B and C were identified based on flow cytometry scatter gates. (D) Absolute lymphocyte counts in the 5 animals on study. Dotted horizontal lines indicate the upper and lower limits of the normal range. LTR, long terminal repeat.
Expansion of gene-modified lymphocytes after in vivo injection of a foamy virus vector. (A) Schematic of the foamy viral vector used in the study. EF1α promoter drives the expression of GFP (to evaluate gene marking) and the coding region of the human common γ chain separated by a 2A self-cleaving peptide. Percentage of lymphocytes expressing (B) γC+ and (C) the GFP transgene in 5 study dogs that received foamy virus in vivo gene therapy, plotted as days postinjection. Lymphocytes in panels B and C were identified based on flow cytometry scatter gates. (D) Absolute lymphocyte counts in the 5 animals on study. Dotted horizontal lines indicate the upper and lower limits of the normal range. LTR, long terminal repeat.
Flow cytometry
Peripheral blood was lysed in hemolytic buffer and washed in phosphate-buffered saline plus 2% fetal bovine serum. Flow cytometry was performed on either an FACSCalibur or FACSCanto flow cytometer (Becton Dickinson, San Jose, CA). Antibodies used are listed in supplemental Methods.
T-cell functional assays
Analysis of T-cell activation and proliferation by measurement of signal transducer and activator of transcription 5 (STAT5) phosphorylation and 5-bromo 2′-deoxyuridine (BrdU) incorporation were performed as previously described.27 Details can be found in supplemental Methods.
TCRVβ spectratyping
Spectratyping was performed as previously described.30 Details can be found in supplemental Methods.
Modified genome sequencing polymerase chain reaction for the detection of retrovirus integration sites
Peripheral blood DNA was isolated using the Gentra Puregene Blood Kit (cat #158908 and #158912; Qiagen, Valencia, CA). Retrovirus integration site amplification, detection, and processing were carried out as previously described, with the exception that random shearing was accomplished by Adaptive Focused Acoustics technology.31-33 Details can be found in supplemental Methods.
Results
Expansion of gene-corrected lymphocytes following intravenous administration of a therapeutic foamy virus vector
Five newborn SCID-X1 dogs received an intravenous injection of foamy virus vector containing the EF1α promoter driving GFP and the coding sequence for the human γC, separated by the 2A self-cleaving peptide (Figure 1A). The genetic background, age at treatment, volume of vector-conditioned medium injected, total dose of infectious units, and survival for each animal are summarized in Table 1. Concentrated foamy virus vector was administered to each dog in 2 fractionated injections, separated by 4 hours, and no acute reaction was observed in any animal following infusion of the virus vector. In all 5 dogs, circulating γC+ lymphocytes could be detected by flow cytometry as early as 2 weeks postinjection. Corrected lymphocytes subsequently expanded in vivo such that γC+ cells comprised as much as 30% to 58% of the total lymphocyte population 12 weeks after injection (Figure 1B). A similar trend was observed for the appearance of GFP+ lymphocytes, with the exception of dog R2162, which failed to develop more than 5% GFP+ lymphocytes before end of study (Figure 1C). In 2 dogs for which long-term follow-up was possible (dog X349 and dog R2202), GFP+ lymphocytes eventually accounted for 73% and 91% of circulating lymphocytes, respectively. With the exception of dog R2162, absolute lymphocyte counts were maintained within normal limits following injection of the therapeutic virus (Figure 1D).
SCID-X1 dogs treated with in vivo–injected foamy virus vector
| Animal | Age at injection | Titer (I.U./mL) | VCM volume (mL) | Final dose (I.U.) | Survival (days) |
|---|---|---|---|---|---|
| X348 | 13 d.o. | 1.4 × 108 | 1.4 (×2) | 4.0 × 108 | 139 |
| X349 | 13 d.o. | 1.4 × 108 | 1.4 (×2) | 4.0 × 108 | 306 |
| R2152 | 12 d.o. | 3 × 108 | 1.4 (×2) | 8.4 × 108 | 101 |
| R2202 | 1 d.o. | 3 × 108 | 0.7 (×2) | 4.2 × 108 | 330 |
| R2203 | 1 d.o. | 3 × 108 | 0.7 (×2) | 4.2 × 108 | 119 |
| Animal | Age at injection | Titer (I.U./mL) | VCM volume (mL) | Final dose (I.U.) | Survival (days) |
|---|---|---|---|---|---|
| X348 | 13 d.o. | 1.4 × 108 | 1.4 (×2) | 4.0 × 108 | 139 |
| X349 | 13 d.o. | 1.4 × 108 | 1.4 (×2) | 4.0 × 108 | 306 |
| R2152 | 12 d.o. | 3 × 108 | 1.4 (×2) | 8.4 × 108 | 101 |
| R2202 | 1 d.o. | 3 × 108 | 0.7 (×2) | 4.2 × 108 | 330 |
| R2203 | 1 d.o. | 3 × 108 | 0.7 (×2) | 4.2 × 108 | 119 |
Virus was delivered in 2 fractionated doses, 4 hours apart, at the day of life indicated for each dog. The VCM volume is volume per injection, and the total volume received is multiplied by 2 for fractionated injections.
d.o., days old; I.U., infectious units; VCM, vector-conditioned medium.
Characterization of T-cell phenotypes
Ectopic expression of γC was sufficient for the development of CD3+ T cells, ranging from 7% to 43% of lymphocytes in the peripheral blood of treated dogs by 6 weeks after administration of therapy (Figure 2A). The majority of CD3+ cells coexpressed GFP that originated from the gene therapy vector and stained positive for CD4 and CD8 (Figure 2B). The majority of CD3+ cells from treated dogs also stained positively for CD45RA, a marker for naïve T cells, indicating recent thymic emigration (Figure 2C).

Expression of T-cell markers on GFP cells. (A) The proportion of CD3+ lymphocytes as a function of days postinjection. (B) GFP+ lymphocytes express T-cell markers CD3 (left), CD4 (middle), and CD8 (right). This representative panel of subset marking was taken from dog X349 at 41 weeks posttreatment. (C) The majority of CD3+ cells stain positive for CD45RA, as indicated in this representative panel of dog X349 at 35 weeks after treatment.
Expression of T-cell markers on GFP cells. (A) The proportion of CD3+ lymphocytes as a function of days postinjection. (B) GFP+ lymphocytes express T-cell markers CD3 (left), CD4 (middle), and CD8 (right). This representative panel of subset marking was taken from dog X349 at 41 weeks posttreatment. (C) The majority of CD3+ cells stain positive for CD45RA, as indicated in this representative panel of dog X349 at 35 weeks after treatment.
The T-cell receptor (TCR) is a heterodimer that interacts with CD3 and either CD4 or CD8 coreceptors on the cell surface and recognizes foreign antigens presented by major histocompatibility complex–class molecules. Mature T cells express 1 of 2 different TCRs and are developmentally classified as either αβ T cells or γδ T cells, according to the specific glycoprotein chains comprising the heterodimer. Most circulating T cells in adult dogs and humans are αβ T cells, whereas γδ T cells commonly reside in the gut mucosa and can respond to foreign pathogens similarly to a pattern-recognition receptor.34 As expected, in an unaffected littermate control, approximately 79.4% and 2.3% of lymphocytes stained positively for the αβ and γδ TCRs, respectively (Figure 3A). We analyzed TCR phenotypes in 2 dogs that received in vivo gene therapy with foamy virus that were followed long-term. In X349, the TCR phenotypes were similar to that of the unaffected control, with 46.2% αβ and 1.5% γδ (Figure 3B). Interestingly, when first analyzed at 22 weeks posttreatment, R2202 exhibited a reversal in the predominant TCR phenotype in circulating lymphocytes, with 16.1% and 49.6% TCR αβ and γδ, respectively (Figure 3C). This reversal in TCR phenotype was maintained throughout the follow-up of this animal, with the percentage of TCRγδ cells in the blood increasing in proportion until the animal was euthanized (Figure 3D).
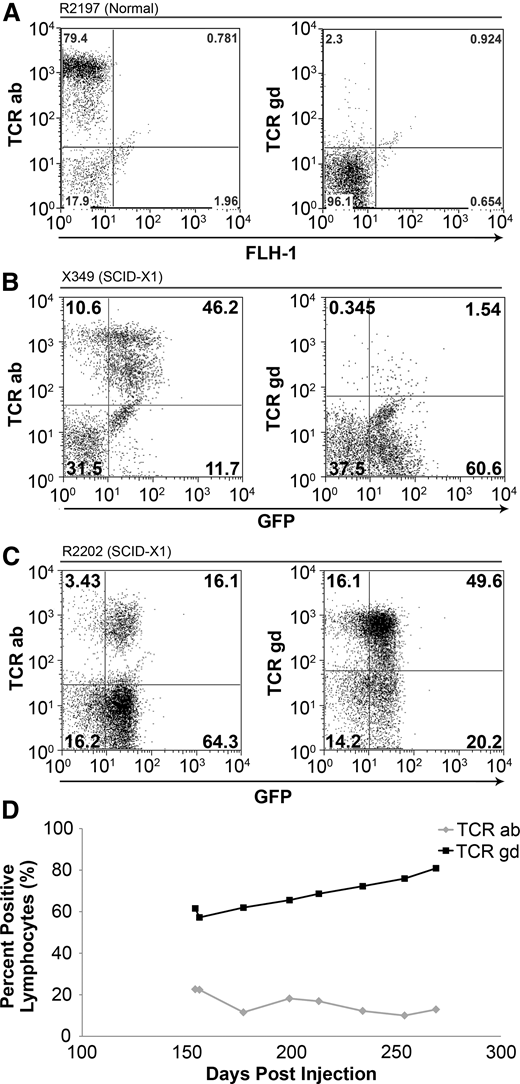
TCR phenotype in foamy-treated dogs. (A) Lymphocytes from an unaffected littermate control (R2197) are approximately 79.4% αβ TCR+ and 2.3% γδ TCR+ at 22 weeks of life. (B) Lymphocytes from X349 at 20 weeks postinjection showed 46.2% αβ TCR+ cells and 1.5% γδ TCR+ cells. (C) R2202 at 28 weeks postinjection exhibited approximately 16.1% αβ TCR+ cells and 49.6% γδ TCR+ cells. (D) The presence of peripheral γδ T cells in R2202 (black line) and αβ T cells (gray line) over time.
TCR phenotype in foamy-treated dogs. (A) Lymphocytes from an unaffected littermate control (R2197) are approximately 79.4% αβ TCR+ and 2.3% γδ TCR+ at 22 weeks of life. (B) Lymphocytes from X349 at 20 weeks postinjection showed 46.2% αβ TCR+ cells and 1.5% γδ TCR+ cells. (C) R2202 at 28 weeks postinjection exhibited approximately 16.1% αβ TCR+ cells and 49.6% γδ TCR+ cells. (D) The presence of peripheral γδ T cells in R2202 (black line) and αβ T cells (gray line) over time.
Rearrangement of the variable region of the TCRβ gene allows T cells to recognize diverse foreign peptides and is one of the earliest T-cell selection events during thymopoiesis.35 We used canine TCRVβ spectratyping to evaluate the T-cell repertoire in SCID-X1 dogs injected with foamy virus.30 In an age-matched littermate control, spectratyping revealed a polyclonal, mostly Gaussian distribution of fragment sizes across 17 families of TCRVβ segments, indicating a diverse TCR repertoire (Figure 4). Dog R2202 displayed mostly polyclonal receptors in nearly all Vβ segments, similar to the TCR repertoire observed in a normal dog, with evidence of oligoclonal TCRVβ rearrangements in some segment families. In contrast, R2203 displayed mostly oligoclonal or skewed (non-Gaussian) distributions and in some cases failed to express TCRVβ segments (indicated as N.D. in Figure 4). At a point close to time of euthanasia (322 d.o.), R2202 lost expression of variable TCR segments. These results demonstrate that delivery of the γC gene via foamy virus in vivo gene therapy facilitates thymocyte development and TCR rearrangement in SCID-X1 dogs.

TCR Vβ spectratyping in foamy virus–treated dogs. The TCR β chain was amplified from complementary DNA using 17 different primer pairs to measure variable region rearrangements. The normal dog (R2197) is a littermate control of R2202 and R2203. Numbers on the left denote the age of the animal at analysis, and the labels at the bottom indicate the forward primer used. d.o., days old; N.D., not detectable. *Sample at necropsy.
TCR Vβ spectratyping in foamy virus–treated dogs. The TCR β chain was amplified from complementary DNA using 17 different primer pairs to measure variable region rearrangements. The normal dog (R2197) is a littermate control of R2202 and R2203. Numbers on the left denote the age of the animal at analysis, and the labels at the bottom indicate the forward primer used. d.o., days old; N.D., not detectable. *Sample at necropsy.
Reconstitution of IL-2 dependent signaling in PBMCs
Stimulation of peripheral blood mononuclear cells (PBMCs) with IL-2 results in downstream tyrosine phosphorylation of STAT5 via activation of the γC chain.36 PBMCs harvested from an unaffected littermate control (R2197) at 48 weeks of age were 31.4% phospho-STAT5 positive after stimulation with recombinant human IL-2 (Figure 5A). At a similar time point, PBMCs from a treated SCID-X1 dog, R2202, displayed approximately 6.3% phospho-STAT5 activation after stimulation (Figure 5B), which represents an activation of approximately 20% that of the unaffected control. In contrast, PBMCs from untreated SCID-X1 dogs fail to display phosphorylation of STAT5 in response to IL-2 stimulation.27
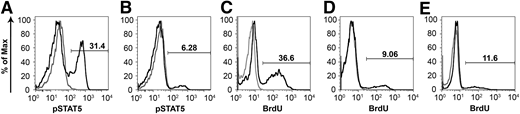
Corrected mononuclear cells activate and proliferate in response to mitogen. PBMCs from (A) an aged-matched normal and (B) SCID-X1 dog R2202 at 46 weeks after injection were treated with recombinant human IL-2, and STAT5 phosphorylation (pSTAT5) was assessed by flow cytometry. PBMCs from the SCID-X1 dog display STAT5 phosphorylation approximately 20% of that of normal. (C-E) Treatment with PHA results in cell-cycle entry of PBMCs derived from a (C) normal unaffected dog or (D-E) foamy virus–treated SCID-X1 dogs X349 at 22 weeks and R2202 at 24 weeks after treatment, respectively. PBMCs proliferate in response to mitogen at ∼25% to 32% of that of the control.
Corrected mononuclear cells activate and proliferate in response to mitogen. PBMCs from (A) an aged-matched normal and (B) SCID-X1 dog R2202 at 46 weeks after injection were treated with recombinant human IL-2, and STAT5 phosphorylation (pSTAT5) was assessed by flow cytometry. PBMCs from the SCID-X1 dog display STAT5 phosphorylation approximately 20% of that of normal. (C-E) Treatment with PHA results in cell-cycle entry of PBMCs derived from a (C) normal unaffected dog or (D-E) foamy virus–treated SCID-X1 dogs X349 at 22 weeks and R2202 at 24 weeks after treatment, respectively. PBMCs proliferate in response to mitogen at ∼25% to 32% of that of the control.
PBMCs from a littermate control and foamy virus–treated SCID-X1 dogs were treated with phytohemagglutinin (PHA), and their ability to re-enter the cell cycle was evaluated by BrdU pulse and flow cytometry. In control cells, stimulation with PHA resulted in approximately 36.6% of PBMCs staining positive for BrdU (Figure 5C). At 22 and 24 weeks postinjection, SCID-X1 dogs receiving gene therapy showed between 9.1% and 11.6% of cells positive for DNA synthesis after stimulation, corresponding to approximately 25% to 32% of the activation observed in a nonaffected control sample, respectively (Figure 5D-E).
Retroviral integration site analysis
Retroviral integration site (RIS) analysis was performed in genomic DNA isolated from leukocytes in 4 of the study animals by modified genomic sequencing polymerase chain reaction (PCR). Early after treatment in dogs X348 and X349 (19 days postinjection), 20 and 36 unique foamy virus RISs were captured, respectively (supplemental Table 1), likely due to low levels of gene marking (<0.1% of PBMCs; Figure 1C). At day 112 postinjection, when gene marking increased (40% in X348 and 38% in X349; Figure 1C), more RISs were captured (113 and 309 RISs, respectively; supplemental Table 1). Based on the identification of ≥20 unique RISs in all samples, all dogs displayed a polyclonal hematopoietic contribution by gene-modified cells over time (supplemental Table 1). However, all dogs demonstrated evidence for dominant clones (RISs constituting ≥20% of all detectable RISs at a given time point) in at least 1 time point analyzed (Figure 6). In dogs X348 and X349, all but 1 of the top 10 most frequently captured RISs were different at each time point (Figure 6B gold asterisk and shaded box). Conversely, in dogs R2202 and R2203, the most frequently captured RISs were common among both time points in both dogs and the most frequently captured RISs constituted 94% and 91% of the total gene-modified cell pool detected in each respective dog at the latest time point analyzed (Figure 6C-D, gold shaded boxes). Based on the frequency of clones in each sample identified by a single sequencing result (“Single capture”), it is unlikely that the pattern of clonal dominance observed was the result of poor sequencing coverage (supplemental Table 1). To rule out PCR bias, we analyzed the number of shear lengths (length of genomic sequence between long terminal repeat junction and linker) observed within dominant RIS-specific sequences. In R2202 and R2203, the respective dominant clones demonstrated an increased shear length representation in addition to increased RIS-specific sequence capture relative to other RISs identified in the sample, indicating true overrepresentation in the original cell pool (supplemental Figure 1).

Longitudinal RIS analysis. All unique RISs identified from (A) X348, (B) X349, (C) R2202, and (D) R2203 are plotted based on the number of times the RIS was sequenced (relative sequence capture) and normalized to the percentage of total RISs captured at each time point for each dog. The gray portion of each bar represents the percentage corresponding to all but the top 10 most frequently captured RISs. The 10 most frequently captured RISs in each sample are represented by the white boxed lower portions of each bar graph. RISs identified at both time points in the same animal were shaded in matching colors in the bar representing each time point. The gold asterisk in panel B represents the position of a clone identified 19 days postinjection that was also sequenced at 112 days postinjection but is impossible to visualize due to the small frequency of this RIS at 19 days postinjection.
Longitudinal RIS analysis. All unique RISs identified from (A) X348, (B) X349, (C) R2202, and (D) R2203 are plotted based on the number of times the RIS was sequenced (relative sequence capture) and normalized to the percentage of total RISs captured at each time point for each dog. The gray portion of each bar represents the percentage corresponding to all but the top 10 most frequently captured RISs. The 10 most frequently captured RISs in each sample are represented by the white boxed lower portions of each bar graph. RISs identified at both time points in the same animal were shaded in matching colors in the bar representing each time point. The gold asterisk in panel B represents the position of a clone identified 19 days postinjection that was also sequenced at 112 days postinjection but is impossible to visualize due to the small frequency of this RIS at 19 days postinjection.
For the top 10 integrations from each time point, identified genomic flanking sequences were aligned to the canine genome (CanFam3.1) using the standalone University of California, Santa Cruz version of BLAT, and the human RefSeq gene closest to the site of virus integration as viewed in the UCSC Genome Browser was used to establish the nearest transcription start site (TSS). Of all top 10 RISs identified (72 unique RISs), 27 (37.5%) were found within a known RefSeq gene-coding region (supplemental Table 2).
Gene marking in myeloid blood lineages and biodistribution of viral integrations
GFP+ cells could be detected in granulocyte populations at low frequencies by flow cytometry (supplemental Figure 2A). GFP+ granulocytes were also detected in R2202 at a frequency of 0.6% using the canine granulocyte marker DM5 (supplemental Figure 2B).37 Thus, in vivo gene therapy can result in the correction of HSCs or myeloid progenitors in addition to circulating common lymphoid progenitors or pro-T lymphocytes, which experience a selective growth advantage after expressing the therapeutic gene.
To evaluate the biodistribution of provirus, we performed RIS analysis on DNA isolated from tissues taken at necropsy from dogs R2202 and R2203. Most integrations sequenced in tissues were also found in peripheral blood samples processed separately, indicating a signal that originated from blood cells in perfused tissue (supplemental Figure 3 and supplemental Table 3). Only 1 RIS found at a high frequency in the gut of R2203 (Chr21:20 269,587; 40.3%) was not sequenced in blood samples from this animal (supplemental Figure 3, red box) and suggests a true off-target transduction event. The 2 RISs sequenced in the ovary of R2202 correspond to the first (93.7%) and third (1.3%) most represented clones in the blood of this animal (121 days postinfusion) and were also discovered in the thymus, lung, and liver, suggesting blood cell contamination. No integrations were sequenced in the testis of R2203. Taken together, intravenous foamy virus gene therapy can result in a low frequency of off-target transduction events, but there is currently no evidence for gene modification of germ-line cells, consistent with that reported for gammaretrovirus in vivo gene therapy in dogs.27
Survival of dogs
The longest period of follow-up was 10.5 months in 2 of the treated dogs (Table 1). Dog X349 was positive by PCR for both cryptosporidium and canine distemper virus, whereas dog R2202 had significant intestinal coccidia on necropsy and was PCR positive for canine distemper virus. Both dogs had been vaccinated with a modified-live vaccine containing canine distemper 6 days (dog R2202) and 4 weeks (dog X349) before being euthanized. Dog R2162 was euthanized 3 months posttreatment and was PCR positive for cryptosporidium. The presence of lymphoid aggregates in the spleen and intestine of this dog suggested early immune reconstitution due to gene therapy. The remaining 2 dogs, R2203 and X348, were euthanized 4 and 4.5 months posttreatment, respectively, with R2203 being PCR positive for canine parainfluenza virus and X348 PCR positive for cryptosporidium. Although all dogs showed evidence of immune reconstitution with functional T cells, infectious complications limited the duration of follow-up (see “Discussion”).
Discussion
Here, we demonstrate that in vivo gene therapy using a therapeutic foamy virus vector can successfully rescue the clinical phenotype of the canine SCID-X1 immunodeficiency. As early as 2 weeks after injection, affected dogs exhibit newly emergent CD3+ T lymphocytes that are mostly GFP positive, which undergo expansion with time in vivo. These T cells display a reasonably even distribution between CD4+ and CD8+ phenotypes, stain positive for CD45RA, and undergo TCRVβ gene rearrangement, indicating active thymopoiesis. Genetically corrected T cells are functionally mature, as demonstrated by their ability to display STAT5 phosphorylation in response to IL-2 and cell-cycle re-entry in response to the T-cell mitogen, PHA.
The time to lymphocyte recovery was slightly longer using a foamy virus vector compared with a previous report using a gammaretrovirus vector injected into 3-day-old SCID-X1 neonates.27 In that study, 3 of 4 dogs exhibited between 64.8% to 69.3% CD3+ lymphocytes 8 weeks after injection, whereas at a similar time point in the current study, 4 of 5 dogs exhibited between 41.7% and 59.7% CD3+ lymphocytes (Figure 2A). The relative lag in recovery time using foamy virus could be attributable to viral titer: dogs treated with gammaretrovirus received 9 to 32 × 108 infectious units, whereas dogs treated with foamy virus received 4 to 8.4 × 108 infectious units. Alternatively, differences in the rate of reconstitution could be due to the relative strengths of the respective retroviral and cellular promoters used. Experiments designed to test dose and promoter efficacy are warranted to achieve an optimal clinical outcome.
Despite normal levels of gene-corrected lymphocytes and demonstration of functional immune reconstitution in 4 of 5 animals, all dogs were euthanized within 1 year of treatment due to enteric and/or respiratory infections. Three dogs were euthanized 3 to 4.5 months posttreatment, suggesting they were exposed to infectious agents during the time when maternal antibody declined. The 2 dogs that survived 10.5 months were vaccinated shortly before their death with a modified-live vaccine. It is unlikely that distemper in these dogs was a result of the attenuated virus, because the incubation period for canine distemper is 10 to 14 days and dog R2202 was vaccinated just 6 days before euthanasia with severe pneumonia and multinucleated giant cells characteristic of canine distemper. Cryptosporidium has recently become a problem in our colony after moving into a newly renovated area. Although drinking water is filtered, affected puppies are exposed to municipal water for kennel cleaning. Antiprotozoan agents, specifically nitazoxanide, are highly effective against cryptosporidium in humans,38,39 and the antibiotic, azithromycin has been demonstrated to manage cryptosporidiosis in dogs.40 Prophylaxis may delay enteric infections and extend the study period.
Intercurrent untreated infections in the dogs likely impact immune values reported in this study, which can confound interpretation of the data. For instance, coccidiosis observed at necropsy may have influenced the proportion of γδ T cells in R2202, which respond to epithelial-tropic pathogens in the gut. Likewise, the loss of TCRVβ diversity in that animal may reflect the skewing toward γδ T cells in this animal at later time points or the selective expansion of some clones in response to persistent infection. Alternatively, these data suggest that a therapeutic threshold was not attained at the dose administered, and subsequent injections or higher infectious titers may have resulted in a different clinical outcome.
RIS analysis on peripheral white blood cells demonstrated a polyclonal contribution, but with clonal dominance observed in all dogs. Dogs R2202 and R2203 exhibited clonal dominance that exceeded 90% contribution to the gene-modified cell pool. Studies in normal dogs using a foamy virus vector expressing GFP and in a canine model of leukocyte adhesion deficiency showed a largely polyclonal repopulation pattern.19,21,22 The SCID model is likely more prone to clonal dominance given the enormous selection pressure for corrected T cells. Another reason for clonal dominance could be the transduction of a limited number of T-cell progenitors, because the single SCID-X1 dog reported as oligoclonal in the gammaretroviral study received the lowest infectious dose.27 Therefore, higher infectious titers or serial infections over several days may increase the number of corrected repopulating cells.
Dog R2202, which exhibited a high percentage of γδ T cells in peripheral blood, displayed a dominant clone mapping to chromosome 4. The closest human RefSeq TSS, RBM34, an RNA-binding motif protein, is >170 kb away from this RIS (supplemental Table 2) and is not implicated in T-cell expansion or associated with TCRγδ expression. R2203 displayed a dominant clone that mapped to chromosome 22, which lies in a poorly annotated region of the canine genome based on the current assembly (canFam3). One RIS in dog R2202 was discovered 993 bp away from the high-mobility group A2 (HMGA2) TSS. Transactivation of HMGA2 by an enhancer element in a lentiviral vector may have contributed to expansion of myeloid cells in a clinical trial for β-thalassemia major.41 Despite the broad tropism of the foamy virus receptor,18 only a single integration in the gut of R2203 (Chr21:20,269,587; 40.3%) appears to result from an off-target transduction. It is very likely, however, that additional off-target events would be discovered at or near the injection site (eg, vascular endothelium), which represents a general risk to intravenous injection of a replication-deficient virus.
The presence of GFP+ granulocytes throughout the length of follow-up suggests that a number of multipotent progenitors with self-renewing capacity are corrected through intravenous in vivo gene therapy. Gene marking in lineages other than T cells is associated with stem cell transduction and long-term reconstitution in SCID patients.11 The discovery of integrations shared between lymphoid and myeloid compartments would indicate the correction of bipotential progenitors; however, low marking (<1%) in the myeloid lineage precluded RIS analysis on sorted populations. On the other hand, we observed a decline in TCR expression in R2202 without a concurrent loss of CD3+ cells at the latest time point (Figures 2A and 4), which could be interpreted as a selection of several T-cell clones with only a moderate repopulation of naïve T cells arising from a limited number of corrected HSCs. Thus, it remains a possibility that transduction of HSCs is limiting at the dosing regimen we used in this study. Alternate delivery methods, timing of injection, and pretreatment regimens can be envisioned that could potentially increase the proportion of multipotent hematopoietic progenitors available for transduction in the bloodstream.
In conclusion, in vivo gene therapy using foamy virus vectors can achieve immune reconstitution in a clinically relevant large animal model of SCID-X1. We did observe clonal dominance, and thus further optimizations in the clinical protocol will be required to explore in more detail the safety profile of foamy virus in vivo gene delivery. Furthermore, differences in early canine and human hematopoiesis, such as significant extramedullary hematopoiesis in the neonatal dog, may have important implications in translating the efficacy of the canine model to a permanent cure for SCID patients. With the advent of newborn screening for SCID42 or the future development of prenatal gene therapy, time to diagnosis can be dramatically shortened to accommodate the most effective window for in vivo human gene replacement. Thus, the development of protocols and reagents to enhance safety and efficacy of in vivo gene therapy is warranted as a potential alternative to current ex vivo approaches in patients.
The online version of this article contains a data supplement.
There is an Inside Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the veterinary staff at the University of Pennsylvania; G. Choi, H. Crawford, and B. Larson for help in preparing the manuscript; A. Adams, D. Chandrasekaran, C. Ironside, K. Norman, and N. Williams for technical assistance; and Dr Linda Burkly of Biogen Idec for providing the γC monoclonal antibody.
This work was supported by National Institutes of Health: National Institute of Allergy and Infectious Diseases, and National Institute of Diabetes and Digestive and Kidney Diseases (grants AI097100 and DK056465). H.-P.K. is a Markey Molecular Medicine Investigator and the recipient of the José Carreras/E.D. Thomas Endowed Chair for Cancer Research.
Authorship
Contribution: C.R.B., B.C.B., G.D.T., A.M.S., T.R.T., D.J.R., P.J.F., and H.-P.K. designed experiments; C.R.B and D.R.K. performed experiments; C.R.B., B.C.B., J.E.A., and G.D.T. analyzed data; C.R.B., B.C.B., and J.E.A. generated figures; M.E.W. generated reagents; and C.R.B., B.C.B., M.E.W., J.E.A., G.D.T., A.M.S., T.R.T., D.J.R., P.J.F., and H.-P.K. wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Hans-Peter Kiem, Fred Hutchinson Cancer Research Center, Mail Stop D1-100, PO Box 19024, Seattle, WA 98109-1024; e-mail: hkiem@fhcrc.org.
