

Abstract
We have previously described critical and nonredundant roles for the phosphoinositide 3-kinase p110δ during the activation and differentiation of naive T cells, and p110δ inhibitors are currently being developed for clinical use. However, to effectively treat established inflammatory or autoimmune diseases, it is important to be able to inhibit previously activated or memory T cells. In this study, using the isoform-selective inhibitor IC87114, we show that sustained p110δ activity is required for interferon-γ production. Moreover, acute inhibition of p110δ inhibits cytokine production and reduces hypersensitivity responses in mice. Whether p110δ played a similar role in human T cells was unknown. Here we show that IC87114 potently blocked T-cell receptor–induced phosphoinositide 3-kinase signaling by both naive and effector/memory human T cells. Importantly, IC87114 reduced cytokine production by memory T cells from healthy and allergic donors and from inflammatory arthritis patients. These studies establish that previously activated memory T cells are at least as sensitive to p110δ inhibition as naive T cells and show that mouse models accurately predict p110δ function in human T cells. There is therefore a strong rationale for p110δ inhibitors to be considered for therapeutic use in T-cell–mediated autoimmune and inflammatory diseases.
Introduction
In many immune-mediated diseases, T cells with an activated or memory phenotype accumulate at the site of tissue destruction. Genetic susceptibility to autoimmunity is often linked to the major histocompatibility complex locus and other loci that affect T-cell biology, thus implying pathologic roles for T cells in autoimmunity.1,2 Indeed, there is mounting evidence that perturbation of antigen receptor signaling in T cells often contributes to autoimmune diseases.3 Therapeutics affecting T cells, such as glucocorticoids, methotrexate, cyclosporine (CS), CTLA4-Ig, and rapamycin, are used effectively to treat or ameliorate immune-related disorders. However, these therapies can be associated with undesirable side effects and/or unresponsiveness in some patients.4-7 Hence, there is a real need for additional drugs that target T cells but do not compromise organ function or leave the patient unduly susceptible to infections.
The class I phosphoinositide 3-kinases (PI3Ks) phosphorylate phosphatidylinositol-(4,5)-phosphate to produce phosphatidylinositol-(3,4,5)-phosphate (PIP3). PIP3 acts as a second messenger by recruiting pleckstrin homology domain-containing proteins to the plasma membrane where they activate signaling pathways that promote proliferation, survival, differentiation, and chemotaxis.8 Class I PI3Ks are subdivided into 2 groups based on their structure: Class IA PI3Ks are heterodimers consisting of one regulatory subunit (p85α, p85β, p50α, p55α, or p55γ) and one catalytic subunit (p110α, p110β, or p110δ), whereas class IB PI3Ks are heterodimers consisting of one regulatory subunit (p101 or p84) and a single catalytic subunit (p110γ).9
p110δ is expressed at high levels in leukocytes and is a major PI3K isoform controlling antigen-evoked immune responses.10,11 Genetic inactivation of p110δ in mice results in impaired B-cell development and function,12-16 diminished primary and secondary T-cell–dependent immune responses,12,17,18 failure of naive cells to differentiate to TH1 or TH2 subsets,19 decreased regulatory T-cell numbers and function,20 and altered antigen-induced trafficking of T cells.21 Nonetheless, T-cell development occurs normally in p110δ-deficient mice, suggesting that p110δ is not essential for all aspects of T-cell receptor (TCR) signaling.12-14 Additional roles for p110δ have been described in mast cells,22,23 neutrophils,24-26 and natural killer cells.27-31 Genetic or pharmacologic inhibition of p110δ using the small-molecule inhibitor IC87114 (IC) reduced disease severity in preclinical rodent models of rheumatoid arthritis,25 asthma,18,32 and allergy,22,23 whereas glucocorticoid resistance was reversed in a smoking-induced airway inflammation model.33 p110δ selective inhibitors also reduced proliferation of acute myeloid leukemia cells and rendered them more sensitive to chemotherapeutics.34,35 Together, these results suggest that small-molecule inhibitors against p110δ may be used to alleviate immune system-mediated diseases. Indeed, p110δ-selective inhibitors are currently being evaluated in phase 1 clinical trials.36
Much of our knowledge of p110δ comes from mouse studies. Although broadly similar, human and mouse immune systems harbor some notable differences.37 This also extends to the PI3K signaling pathway because human neutrophils are more reliant on p110δ than mouse neutrophils for formyl-methionyl-leucyl-phenylalanine responses.24 This result stresses the requirement to validate results obtained from mouse studies in human experimental model systems. It is also worth noting that, as a consequence of multiple exposures to infectious organisms, humans tend to have greater proportions of memory cells than do mice bred in a pathogen-free environment. Inhibiting previously activated T cells will probably be critical if p110δ inhibitors are to be used effectively to treat T cell–mediated pathologies.
In the p110δD910A mouse, which was genetically engineered to express catalytically inactive p110δ, naive CD4+ T cells proliferated poorly and produced significantly less cytokine, suggesting that p110δ activity is required for optimal responses to antigen, especially under suboptimal stimulation conditions (eg, in the absence of costimulation).12,17,19 Previously activated T cells (referred to here as effector or memory T cells) have a lower threshold of activation linked to rewiring of signal transduction pathways and are less dependent on costimulation.38 Hence, it was not obvious whether p110δ would boost activation of memory T cells as it does in naive T cells. In this study, we have used IC to acutely inhibit p110δ in effector and memory T cells from mice and humans and monitored intracellular signaling events, proliferation, cytokine production, and inflammatory responses.
We found that sustained p110δ activation is required for interferon-γ (IFN-γ) production and that IC potently blocked effector responses from effector/memory T cells in both species. p110δ inhibition also reduced contact-hypersensitivity (CHS) reactions in mice, and cytokine production by T cells from patients with allergy or reactive arthritis (ReA). We conclude that p110δ plays an important role in the activation of effector and memory T cells in humans and mice.
Methods
Mice
p110δD910A, OT1, and OT2 mice have been described previously,12,39,40 and were backcrossed to C57BL/6 background for at least 10 generations. Mice were maintained under specific pathogen–free conditions. All protocols involving live animals were approved by the United Kingdom Home Office and institutional ethics review.
Reagents
IC8711426 was synthesized as described (D030 from patent WO 01/81346). IC inhibits p110δ kinase activity with an IC50 between 0.1 and 0.5μM, and only shows cross-reactivity with other PI3K isoforms at concentrations more than 10μM.26,41 CS was synthesized at UCB Pharma. OVA257-264 and OVA323-339 were synthesized by Southampton Polypeptides. Other chemicals were purchased from Sigma-Aldrich unless otherwise specified.
Culture media
Mouse T cells were cultured in RPMI 1640 (Invitrogen) with 5% fetal calf serum (TCS Cellworks), 1% penicillin-streptomycin, and β-mercaptoethanol. Human cells were cultured in RPMI 1640 and 1% penicillin-streptomycin and either 5% human AB serum or 10% fetal calf serum. Cells were incubated at 37°C with 5% CO2.
Mouse cell purification and activation
CD4+ or CD8+ T cells were negatively selected from lymph nodes and/or spleens by magnetic sorting. Cells were incubated with fluorescein isothiocyanate–labeled anti-B220, -CD25, -CD69, -CD49b, and –major histocompatibility complex class II antibodies. CD8+ T-cell purifications also included anti-CD4, whereas CD4+ T-cell purifications included anti-CD8 (all eBioscience). Antibody-coated cells were then depleted with anti–fluorescein isothiocyanate MACS beads (Miltenyi Biotec). The resulting population was more than 95% CD4+ or CD8+. For naive and effector/memory T-cell studies, negatively selected CD4+ T cells were stained with anti-CD62L–PE (eBioscience) before being sorted by fluorescence-activated cell sorter into CD62L+ and CD62L− populations. The resulting populations were greater than 97% pure.
To activate CD8+ OT1 T cells or CD4+ OT2 T cells, purified cells were stimulated in triplicate in a 1:1 ratio with irradiated T cell–depleted spleen cells pulsed with 1μM chicken ovalbumin peptide (OT1:OVA257-264 peptide; OT2: OVA323-339 peptide) in the presence of dimethyl sulfoxide (DMSO) alone, LY294002 (LY), or IC. Polyclonal CD62L+ and CD62L− CD4+ T cells were activated in triplicate with 1 μg/mL plate-bound anti-CD3 (clone 2C11) in the presence of DMSO alone, LY, or IC. Supernatants were collected at 48 hours and assayed by enzyme-linked immunosorbent assay (eBioscience), and cells were pulsed with 3H-thymidine overnight before harvesting.
Anti-CD3 and ConA injections
Mice were dosed once orally with 30 mg/kg IC in 1% methylcellulose (MCL) or with 1% MCL vehicle control alone 30 minutes before they were injected intravenously with anti-CD3 (0.01 mg/kg) or concanavalin A (ConA; 15 mg/kg). At 1.5 hours after injection, the mice were terminally anesthetized and bled by heart puncture. Cytokine levels in the serum were measured using a FlowCytomix kit from Bender Medsystems.
CHS studies
A total of 100 μL of 7% trinitrochlorobenzene (TNCB) in a 4:1 acetone/olive oil mix or acetone/olive oil alone (for unsensitized controls) was applied to the shaved abdomens of p110δD910A or C57BL/6J controls. Six days later, ear thickness was measured with a micrometer (Kroeplin). Mice were subsequently dosed orally with 30 mg/kg IC in 1% MCL or with 1% MCL vehicle control alone 1 hour before and 5 hours after elicitation of CHS. To elicit CHS, 1% TNCB in a 9:1 olive oil/acetone mix was applied to an ear. Ear thickness was measured again 24 hours later. The change in ear thickness was measured as the size difference before and after rechallenge.
Human cell purification
Peripheral blood mononuclear cells (PBMCs) from healthy humans were isolated using a Ficoll gradient. In some experiments, cells were negatively selected according to the manufacturer's instructions using RosetteSep Human T cell Enrichment Cocktail (for total T cells), or EasySep Human Naive CD4+ T cell or Memory CD4+ T cell Enrichment kits (for naive or effector/memory T cells, respectively) (Stem Cell Technologies). Total CD4+ T cells were negatively selected from atopic donors using Human CD4+ T Cell Isolation Kit II (Miltenyi), and autologous antigen-presenting cells (APCs) isolated from the positively selected fraction. Purified cells were more than 95% pure. Mononuclear cells were also isolated from the synovial fluid of patients fulfilling European Spondylarthropathy Study Group criteria for spondyloarthritis, and had clinical ReA.42 All human tissues had been donated under informed consent in accordance with the Declaration of Helsinki, and experiments were done in accordance with institutional ethical review guidelines.
Human T-cell biochemistry
For Western blot analysis, total (CD3+) T cells were pretreated with DMSO alone, LY, or IC and subsequently stimulated by first adding 0.3 μg/mL anti-CD3 (clone OKT3) and 1 μg/mL anti-CD28 (clone CD28.2; Immunotech). After 2 minutes, these antibodies were cross-linked for 5 minutes with 10 μg/mL goat anti–mouse IgG, F(ab′)2 (Jackson ImmunoResearch). Lysates were probed with polyclonal rabbit anti–phospho-Ser473-Akt, -Thr308-Akt, -Thr202/Tyr204-Erk1/2, total Akt, and total Erk1/2 (all Cell Signaling Technology).
For fluorescent cell bar-coding experiments, total T cells were pretreated with DMSO alone, LY, or IC and subsequently stimulated by first adding 0.3 μg/mL biotinylated anti-CD3 and 5 μg/mL biotinylated anti-CD28. After 2 minutes, cells were cross-linked with 20 μg/mL avidin (Zymed) at 37°C, and aliquots were withdrawn at the indicated time points. Cells were fixed for 10 minutes at 37°C with Fix Buffer I (BD Biosciences), washed, and then fluorescently bar-coded as described previously.43 Briefly, aliquots were stained for 20 minutes at room temperature in different dilutions of AlexaFluor 488 and Pacific Blue succinimidyl ester (Invitrogen), providing each sample with a unique fluorescent signature. Cells were washed and then pooled for further staining. Cells were permeabilized in Perm Buffer III (BD Biosciences), washed, and stained with phospho-specific antibodies (Cell Signaling Technology): Alexa 647–conjugated pSer473-Akt clone D9E, pThr202/Tyr204-Erk1/2 clone E10, or pSer235/236-S6 clone D57.2.2E; or unconjugated pSer9-GSK3β followed by Alexa 647 goat anti–rabbit IgG (Invitrogen) and lineage markers (CD3 clone SK7, CD4 clone SK3, CD45RO clone UCHL1; all BD Biosciences) for 30 minutes at room temperature. T-cell subsets were defined as follows: naive CD4+ (CD3+CD4+CD45RO−), effector/memory CD4+ (CD3+CD4+CD45RO+), naive CD8+ (CD3+CD4−CD45RO−), and effector/memory CD8+ (CD3+CD4−CD45RO+). Cells were processed on a FACSCanto II flow cytometer (BD Biosciences). Data were analyzed using the Stanford University Cytobank program (www.cytobank.stanford.edu). Arcsinh medians with a cofactor of 150 were used for the statistical calculation of fold change in Alexa 647 between individual samples using the following equation:
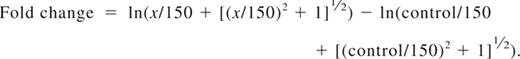
Results were visualized either as heatmaps or as overlaid histograms.
Human T-cell activation
CD3+ human primary T cells were activated with T Cell Expander Dynabeads (Invitrogen) in a 1:1 bead-to-cell ratio. Supernatants for interleukin-2 (IL-2) and IFN-γ measurements were collected after 16 hours. CD45RA+ or CD45RO+ CD4+ T cells were stimulated with 3.75 μg/mL plate-bound anti-CD3 (Clone UCHT1; BD Biosciences) for 48 hours before measuring proliferation and cytokine production. Total PBMCs were stimulated with 1 μg/mL tetanus toxin (Calbiochem) for 7 days before measuring proliferation and cytokine production. CD4+ T cells from PBMCs of atopic donors were mixed 1:1 with autologous APCs and stimulated with 25 μg/mL house dust mite antigen (Greer Laboratories) for 6 days before measuring cytokine production. Mononuclear cells from the synovial fluid of ReA patients presenting in clinic were stimulated with plate-bound anti-CD3 for 72 hours before measuring proliferation and cytokine production. Cytokine concentrations from culture supernatants were assayed by enzyme-linked immunosorbent assays from R&D Systems or MesoScale Discovery.
Statistics
In mouse experiments, the effects of genetic or pharmacologic inactivation of p110δ were compared with uninhibited wild-type (WT) controls in mice using Student t tests, except for the CHS studies where the 1-way analysis of variance with a Bonferroni posttest was used. Responses of human cells treated with IC were compared with DMSO controls using 1-sided repeated measures analysis of variance on log-transformed data. Responses of cells treated with LY or CS were compared with DMSO controls using Student t test on log-transformed data. Probabilities were calculated using GraphPad Prism or SPSS. Percentage inhibition of human cells was calculated as 100 × (1 − [inhibited sample − unstimulated sample]/[uninhibited sample − unstimulated sample]), and median values are shown below the graphs, with negative numbers indicating an enhanced response.
Results
Naive and effector/memory mouse CD4+ T cells are sensitive to p110δ inhibition
We have previously shown that p110δ is required for PIP3 accumulation for at least 30 minutes after TCR stimulation in CD4+ T cells.17 However, the kinetics of PI3K activation after this time point is unknown. CD8+ T cells only required PI3K activation for the first 9 hours after activation to induce a full proliferative program.44 This suggested that prolonged activation of PI3Ks (including p110δ) may not be required in T cells.
To examine this hypothesis further, OT1 (CD8+) and OT2 (CD4+) TCR transgenic cells were activated with APCs and peptide, and either 10μM of the pan-PI3K inhibitor LY or 5μM of the p110δ-selective inhibitor IC was added at the start of the culture or up to 24 hours later. Proliferation of IL-2 and IFN-γ production after 48 hours were subsequently compared with DMSO-treated controls. In both CD8+ and CD4+ T cells, proliferation and IL-2 production were only minimally affected when either LY or IC was added 24 hours after activation (Figure 1A-D). By contrast, delayed addition of IC profoundly blocked IFN-γ production (Figure 1E-F). We conclude that, both in CD4+ and CD8+ T cells, p110δ activity needs to be sustained for longer to optimize IFN-γ production than is required for proliferation or IL-2 production.

p110δ regulates cytokine production late after TCR stimulation. CD8+ OT1 (left column) or CD4+ OT2 (right column) T cells were stimulated with irradiated APCs plus 1mM OVA peptide or left unstimulated. A total of 10mM LY or 5mM IC was added at the start of coculture or 7 or 24 hours later. Vehicle control (DMSO) was added at the experiment start for uninhibited controls. After 48 hours, proliferation (A-B), IL-2 (C-D), and IFN-γ production (E-F) were measured. Data are mean ± SEM of triplicate readings and are representative of 2 independent experiments.
p110δ regulates cytokine production late after TCR stimulation. CD8+ OT1 (left column) or CD4+ OT2 (right column) T cells were stimulated with irradiated APCs plus 1mM OVA peptide or left unstimulated. A total of 10mM LY or 5mM IC was added at the start of coculture or 7 or 24 hours later. Vehicle control (DMSO) was added at the experiment start for uninhibited controls. After 48 hours, proliferation (A-B), IL-2 (C-D), and IFN-γ production (E-F) were measured. Data are mean ± SEM of triplicate readings and are representative of 2 independent experiments.
We next asked whether previously activated CD4+ T cells also retained sensitivity to p110δ inhibition. CD4+ T cells from WT and p110δD910A mice were sorted into naive CD62L+ and effector/memory CD62L− populations and stimulated with plate-bound anti-CD3 in the presence of DMSO, LY, or IC added for the duration of the experiment. Proliferation was blocked in WT CD62L+ and CD62L− cells with an IC50 of 1.233 plus or minus 0.16μM and of 0.04 plus or minus 0.03μM, respectively (Figure 2A,C). IFN-γ production was 10-fold more sensitive with IC50 of 0.12 plus or minus 0.09μM and 0.001 plus or minus 0.0007μM in WT CD62L+ and CD62L− cells, respectively (Figure 2B,D). Inactivation of p110δ by 1μM IC blocked cytokine responses to similar levels as did 10μM LY (Figure 2E-F). Naive CD62L+ CD4+ T cells from p110δD910A mice proliferated poorly and produced only 20% of the IFN-γ seen in WT cells (Figure 2A-B) as seen previously.19 Here we extend these findings by showing that the same pattern persists in previously activated CD62L− p110δD910A T cells. Although IC showed some suppression of p110δD910A T-cell proliferation at the higher concentrations, the low amount of IFN-γ produced by p110δD910A T cells was not further reduced by IC (compare Figure 2A,C with Figure 2B,D). The minimal impact of IC on IFN-γ production by p110δD910A cells suggests that the inhibition of WT cells at low concentrations was not the result of off-target effects (Figure 2B,D).

p110δ regulates naive and effector/memory CD4+ T-cell activation. CD62L+ (naive) and CD62L− (effector/memory) CD4+ T cells from WT and p110δD910A mice were stimulated by anti-CD3 in the presence of DMSO alone, LY, or IC. After 48 hours, proliferation (A,C,E) and IFN-γ production (B,D,F) were measured. The effects of IC on naive (A-B) or effector/memory (C-D) T cells were determined. Proliferation (E) and IFN-γ production (F) were measured in WT cells when cells were treated with DMSO alone, 10mM LY, or 1mM IC. Data are mean ± SEM of replicate readings and are representative of at least 2 independent experiments.
p110δ regulates naive and effector/memory CD4+ T-cell activation. CD62L+ (naive) and CD62L− (effector/memory) CD4+ T cells from WT and p110δD910A mice were stimulated by anti-CD3 in the presence of DMSO alone, LY, or IC. After 48 hours, proliferation (A,C,E) and IFN-γ production (B,D,F) were measured. The effects of IC on naive (A-B) or effector/memory (C-D) T cells were determined. Proliferation (E) and IFN-γ production (F) were measured in WT cells when cells were treated with DMSO alone, 10mM LY, or 1mM IC. Data are mean ± SEM of replicate readings and are representative of at least 2 independent experiments.
To determine the effect of acute p110δ inhibition on cytokine responses in vivo, mice were injected with anti-CD3 or ConA and cytokine levels in the serum were measured 1.5 hours later. Oral administration of IC reduced IL-2, IL-4, IL-17, IFN-γ, and tumor necrosis factor-α (TNF-α) levels in the serum (Figure 3A-E). These results demonstrate that IC can block T-cell–dependent cytokine production in vivo.

IC reduces T-cell cytokine production and contact hypersensitivity responses in vivo. WT B6 mice were given one oral dose of IC (30 mg/kg) 30 minutes before being administered anti-CD3 or ConA intravenously. Cytokine concentrations in the serum 1.5 hours after injection were measured: (A) IL-2, (B) IL-4, (C) IL-17, (D) IFN-γ, and (E) TNF-α. (F-G) WT and p110δD910A mice were sensitized on the abdomen with TNCB and then rechallenged with TNCB on one ear 6 days (F; primary response) or 6 and 30 days (G; secondary response) later. Mice were dosed twice orally with 30 mg/kg IC or MCL alone on the day of the final rechallenge, and the change in ear thickness was measured 24 hours later. The effect of p110δ genetic or pharmacologic inactivation was compared with WT mice receiving MCL only, and P values were calculated using 1-way analysis of variance with Bonferroni posttest. *.01 < P < .05; **.001 < P < .01; ***P < .001. Data are representative of 3 experiments for primary responses and 1 experiment for secondary responses.
IC reduces T-cell cytokine production and contact hypersensitivity responses in vivo. WT B6 mice were given one oral dose of IC (30 mg/kg) 30 minutes before being administered anti-CD3 or ConA intravenously. Cytokine concentrations in the serum 1.5 hours after injection were measured: (A) IL-2, (B) IL-4, (C) IL-17, (D) IFN-γ, and (E) TNF-α. (F-G) WT and p110δD910A mice were sensitized on the abdomen with TNCB and then rechallenged with TNCB on one ear 6 days (F; primary response) or 6 and 30 days (G; secondary response) later. Mice were dosed twice orally with 30 mg/kg IC or MCL alone on the day of the final rechallenge, and the change in ear thickness was measured 24 hours later. The effect of p110δ genetic or pharmacologic inactivation was compared with WT mice receiving MCL only, and P values were calculated using 1-way analysis of variance with Bonferroni posttest. *.01 < P < .05; **.001 < P < .01; ***P < .001. Data are representative of 3 experiments for primary responses and 1 experiment for secondary responses.
To test whether p110δ inhibition could impair functional recall responses in vivo, the ability of IC to block elicitation of a T-cell–dependent CHS reaction was tested. WT or p110δD910A mice were sensitized topically on their abdomens with the hapten TNCB and then rechallenged after 6 days on one ear. Mice were dosed twice with either 30 mg/kg IC or 1% methylcellulose (MCL) vehicle control on the day of rechallenge, and ear swelling was measured 24 hours later (Figure 3F). Ear swelling was reduced by approximately 60% in both WT mice treated with IC and in p110δD910A mice, showing that acute blockade of p110δ in primed cells reduced hypersensitivity as effectively as blocking p110δ chronically in naive cells. IC treatment in p110δD910A mice did not suppress swelling further, showing that inhibition by drug was not the result of off-target effects (Figure 3F). No response was observed from either genotype when mice were challenged on their ear in the absence of prior sensitization (data not shown), confirming that the swelling responses were mediated by primed effector cells.
To determine whether a secondary response to TNCB retained the same sensitivity to p110δ inhibition, WT and p110δD910A mice were sensitized and elicited as before except without drug treatment. Four weeks after initial sensitization, ears were TNCB-challenged for a second time and mice were dosed twice with either 30 mg/kg IC or 1% MCL vehicle control on the day of the second rechallenge. The ears of both WT and p110δD910A mice were more swollen after the second elicitation than the first (Figure 3G; and data not shown), indicating that a memory-type response was being measured. Similar to the primary response (Figure 3F), an approximate 50% reduction in swelling was observed in IC-treated WT mice as well as in p110δD910A mice with or without IC treatment (Figure 3G). We conclude that p110δ participates in primary and secondary responses in vivo.
p110δ is the main transducer of PI3K signals in TCR-stimulated human T cells
We have shown that TCR-induced phosphorylation of Akt in mouse T cells is highly sensitive to p110δ inhibition.12,17,19 Because Akt phosphorylation only occurs if PIP3 is produced, these results demonstrated that p110δ is the main PI3K isoform activated by the TCR. To determine whether human T cells were also dependent on p110δ to activate the PI3K pathway, primary T cells from healthy donors were stimulated for 5 minutes with anti-CD3 and anti-CD28 in the presence of DMSO alone, IC, or LY, and phosphorylation of Akt and Erk were subsequently analyzed by immunoblotting (Figure 4A). Both LY and IC blocked Akt phosphorylation in a dose-dependent manner. IC inhibited Akt phosphorylation as potently as LY, suggesting that p110δ transmits PI3K signals from the TCR and CD28 in human T cells and that other isoforms cannot substantially compensate. Erk phosphorylation was only partially blocked by 10μM of either inhibitor, consistent with a nonessential but accessory role for PI3K in regulating Erk in T cells under some stimulatory conditions.12,19

p110δ regulates TCR signaling by human T cells. (A) T cells from healthy human donors were pretreated with DMSO alone, IC, or LY and stimulated with anti-CD3 and anti-CD28 for 5 minutes. Lysates were immunoblotted to examine the levels of total and phosphorylated Akt and Erk. Data represent one of 3 donors. (B-F) T cells from healthy human donors were pretreated with DMSO alone, IC, or LY and stimulated with anti-CD3 and anti-CD28 for different time points. The pooled bar-coded aliquots were stained for surface markers and phospho-proteins and analyzed by flow cytometry. Populations were gated into naive (N) and effector/memory (E/M) CD4+ and CD8 + T cells, and changes in phosphorylation were assessed for each population. Histograms showing dynamics of phosphorylation of Akt (B), GSK3β (C), Erk (D), and S6 (E) in DMSO-treated samples are shown. (F) The data for DMSO- and inhibitor-treated samples were converted into a heatmap showing fold change compared with unstimulated control. The data presented are representative of 3 independent experiments with different donors.
p110δ regulates TCR signaling by human T cells. (A) T cells from healthy human donors were pretreated with DMSO alone, IC, or LY and stimulated with anti-CD3 and anti-CD28 for 5 minutes. Lysates were immunoblotted to examine the levels of total and phosphorylated Akt and Erk. Data represent one of 3 donors. (B-F) T cells from healthy human donors were pretreated with DMSO alone, IC, or LY and stimulated with anti-CD3 and anti-CD28 for different time points. The pooled bar-coded aliquots were stained for surface markers and phospho-proteins and analyzed by flow cytometry. Populations were gated into naive (N) and effector/memory (E/M) CD4+ and CD8 + T cells, and changes in phosphorylation were assessed for each population. Histograms showing dynamics of phosphorylation of Akt (B), GSK3β (C), Erk (D), and S6 (E) in DMSO-treated samples are shown. (F) The data for DMSO- and inhibitor-treated samples were converted into a heatmap showing fold change compared with unstimulated control. The data presented are representative of 3 independent experiments with different donors.
Some signaling differences have been observed in naive, activated, and memory T cells, and between CD4+ and CD8+ T cells.38,45,46 Hence, it was plausible that IC may affect T-cell subsets differently. To compare the potency of IC on different T-cell subsets, purified primary T cells were treated with DMSO alone, IC, or LY before stimulation with anti-CD3 and anti-CD28, and PI3K targets were analyzed by phospho-flow cytometry at various time points (Figure 4B-F). By analyzing populations separately on the basis of surface phenotype, the effects of inhibitors could be assessed in naive CD4+ (CD3+CD4+CD45RO−), effector/memory CD4+ (CD3+CD4+CD45RO+), naive CD8+ (CD3+CD4−CD45RO−), and effector/memory CD8+ T cells (CD3+CD4−CD45RO+). Raw data were then converted into a heatmap comparing the change in fluorescence to an unstimulated control. Histograms for samples treated with DMSO alone (Figure 4B-E) are shown to demonstrate the levels of phosphorylation represented by each color in the heatmap (Figure 4F). Each subset reacted similarly to IC, with phosphorylation of Akt, S6, and GSK3β reduced at even the lowest concentration of IC, whereas Erk phosphorylation was only diminished at 10μM IC (Figure 4F). These data demonstrate that, in each T-cell subset, CD3 and CD28 activate p110δ and that prior activation status does not influence sensitivity to IC. Moreover, these data reveal redundancy in signals leading to GSK3 and S6 phosphorylation as these phosphorylation events were less sensitive to PI3K inhibition than was Akt phosphorylation. Indeed, Erk and Rsk are known to contribute to these signaling events independently of PI3K.47,48
Because IFN-γ is primarily secreted by previously activated T cells, we next subfractionated CD4+ T cells isolated from healthy donors into naive CD45RA+ and effector/memory CD45RO+ populations, and stimulated for 48 hours with anti-CD3 in the presence DMSO alone, LY, or IC. Up-regulation of the activation marker CD69 was undiminished in all but the highest concentration of PI3K inhibitors, which correlated with the level of Erk inhibition (Figure 5A-B). However, IC concentrations between 1 and 10μM diminished proliferation of both CD45RA+ and CD45RO+ CD4+ T cells to a similar extent as LY (Figure 5C-D). IFN-γ and IL-17 production was undetectable in CD45RA+ cells from most donors, but LY and IC profoundly blocked production of IFN-γ and IL-17 by CD45RO+ cells (Figure 5F,H). Individual donors varied in sensitivity to IC at the lowest concentration used (0.1μM). To indicate the most common level of inhibition observed, percentage inhibition was calculated for each donor as described in “Statistics” and the median value shown beneath the graph. These data suggest that human effector/memory T cells are at least as sensitive to p110δ inhibition as naive T cells.

p110δ inhibitors block proliferation and cytokine production by naive and effector/memory CD4+ T cells. CD4+CD45RA+ (left column) and CD4+CD45RO+ (right column) T cells were stimulated with anti-CD3 in the presence of DMSO alone, IC, or LY. At 48 hours, CD69 expression (A-B), proliferation (C-D), IFN-γ (E-F), and IL-17 (G-H) production were measured. (A-B) Data are representative of one of 3 donors. (C-H) Each dot represents an individual donor. To calculate P values, Student t test was used to compare DMSO alone with LY, and 1-way repeated measures analysis of variance was used to compare DMSO alone with IC. *.01 < P < .05; **.001 < P < .01; ***P < .001. Percentage inhibition was calculated for each donor to determine the range of sensitivity in the population, and median values are shown below the graph. (I-F) T cells were stimulated for 16 hours with anti-CD3 and anti-CD28 in the presence of DMSO alone, IC, or LY, and IL-2 (I) or IFN-γ (J) production was measured. Data are mean ± SEM of triplicate readings and are representative of 1 of 3 donors.
p110δ inhibitors block proliferation and cytokine production by naive and effector/memory CD4+ T cells. CD4+CD45RA+ (left column) and CD4+CD45RO+ (right column) T cells were stimulated with anti-CD3 in the presence of DMSO alone, IC, or LY. At 48 hours, CD69 expression (A-B), proliferation (C-D), IFN-γ (E-F), and IL-17 (G-H) production were measured. (A-B) Data are representative of one of 3 donors. (C-H) Each dot represents an individual donor. To calculate P values, Student t test was used to compare DMSO alone with LY, and 1-way repeated measures analysis of variance was used to compare DMSO alone with IC. *.01 < P < .05; **.001 < P < .01; ***P < .001. Percentage inhibition was calculated for each donor to determine the range of sensitivity in the population, and median values are shown below the graph. (I-F) T cells were stimulated for 16 hours with anti-CD3 and anti-CD28 in the presence of DMSO alone, IC, or LY, and IL-2 (I) or IFN-γ (J) production was measured. Data are mean ± SEM of triplicate readings and are representative of 1 of 3 donors.
Because CD28 costimulation has been shown, under some circumstances, to be insensitive to p110δ inhibition,12 we also stimulated human T cells with anti-CD3 and anti-CD28. Consistent with previous results in mouse,12 we found that anti-CD28 stimulated robust IL-2 production in the absence of p110δ activity. However, IFN-γ production was inhibited by IC in a dose-dependent manner. The ability of LY, but not IC to inhibit IL-2 production, may reflect a role for mTOR in blocking IL-2 production as LY, but not IC, targets the catalytic activity of this kinase.41
CD45R0+ cells include both recently activated effector and resting memory cells. As a second, more definitive approach to monitor the sensitivity of human memory T cells to p110δ inhibition, unfractionated PBMCs were pulsed with tetanus toxin in the presence of DMSO alone or IC. Tetanus toxin will only elicit a response from persons who have previously been vaccinated against tetanus and the response is mediated by memory cells. Proliferation was unaffected by p110δ in this context (Figure 6A, although we observed some inhibition of proliferation in other experiments. However, T cell–derived cytokine production was still blocked by IC, with more than 50% of IFN-γ and TNF-α production blocked at more than 1μM and IL-17 blocked at less than 0.1μM (Figure 6B-D). We conclude that cytokine production by human memory T cells is highly sensitive to p110δ inhibition.

p110δ inhibition blocks recall responses. PBMCs from healthy donors were stimulated with tetanus toxin in the presence of DMSO alone or IC. After 7 days, proliferation (A), IFN-γ (B), TNF-α (C), and IL-17 (D) production were measured. Each dot represents an individual donor. To calculate P values, 1-way repeated-measures analysis of variance was used to compare DMSO alone with IC. *.01 < P < .05; **.001 < P < .01; ***P < .001. Percentage inhibition was calculated for each donor to determine the range of sensitivity in the population, and median values are shown below the graph.
p110δ inhibition blocks recall responses. PBMCs from healthy donors were stimulated with tetanus toxin in the presence of DMSO alone or IC. After 7 days, proliferation (A), IFN-γ (B), TNF-α (C), and IL-17 (D) production were measured. Each dot represents an individual donor. To calculate P values, 1-way repeated-measures analysis of variance was used to compare DMSO alone with IC. *.01 < P < .05; **.001 < P < .01; ***P < .001. Percentage inhibition was calculated for each donor to determine the range of sensitivity in the population, and median values are shown below the graph.
p110δ inhibitors block cytokine production from disease-associated T cells
Because p110δ inhibitors are being developed to treat immune disorders, we asked how effective IC was in dampening T-cell responses from patients presenting with atopy or inflammatory arthritis. Unfractionated PBMCs from allergic persons were stimulated with house dust mite antigen in the presence of DMSO alone or IC, and assayed after 6 days for TH2 cytokine production (Figure 7A-B). In 5 donors, IC blocked cytokine production potently, although a sixth donor was unresponsive. IC blocked IL-5 production with an IC50 range between 0.06 and 0.35μM and IL-13 with an IC50 range between 0.1 and 0.3μM (data not shown). Our data show that allergen-driven TH2 T cells are sensitive to p110δ inhibition.

p110δ inhibition blocks recall responses from disease samples. (A-B) PBMCs from atopic persons were pulsed with house dust mite antigen in the presence of DMSO alone or IC. After 6 days, IL-5 (A) and IL-13 (B) were measured. (C-F) Mononuclear cells from synovial fluid of ReA patients were stimulated with anti-CD3 in the presence of DMSO alone, IC, or CS. Proliferation (C), IFN-γ (D), IL-17 (E), and TNF-α (F) production were measured at 72 hours. Each dot represents an individual donor. To calculate P values, Student t test was used to compare DMSO alone with CS, and 1-way repeated-measures analysis of variance was used to compare DMSO alone with IC. *.01 < P < .05; **.001 < P < .01; ***P < .001. Percentage inhibition was calculated for each donor to determine the range of sensitivity in the population, and median values are shown below the graph.
p110δ inhibition blocks recall responses from disease samples. (A-B) PBMCs from atopic persons were pulsed with house dust mite antigen in the presence of DMSO alone or IC. After 6 days, IL-5 (A) and IL-13 (B) were measured. (C-F) Mononuclear cells from synovial fluid of ReA patients were stimulated with anti-CD3 in the presence of DMSO alone, IC, or CS. Proliferation (C), IFN-γ (D), IL-17 (E), and TNF-α (F) production were measured at 72 hours. Each dot represents an individual donor. To calculate P values, Student t test was used to compare DMSO alone with CS, and 1-way repeated-measures analysis of variance was used to compare DMSO alone with IC. *.01 < P < .05; **.001 < P < .01; ***P < .001. Percentage inhibition was calculated for each donor to determine the range of sensitivity in the population, and median values are shown below the graph.
As in rheumatoid arthritis, T cells from the synovial fluid of ReA patients have an effector/memory phenotype and secrete large quantities of IL-17 and TNF-α.49 We stimulated ReA synovial fluid mononuclear cells with anti-CD3 in the presence of DMSO alone, IC, or CS, and proliferation and arthritis-associated cytokine production were assayed after 72 hours. Proliferation was unaffected, but production of IFN-γ, IL-17, and TNF-α was blocked in a dose-dependent manner with the highest concentrations of IC achieving a similar abrogation as CS (Figure 7C-F). This suggests that p110δ inhibition has the potential to be an effective blocker of TH1 and TH17 inflammatory T-cell responses in the context of inflammatory arthritis.
Discussion
In this study, we have demonstrated that both mouse and human effector/memory T cells are highly sensitive to p110δ inhibition. Moreover, cytokine production by disease-enhancing T cells can be blocked by a p110δ-specific inhibitor.
In mice, genetic models showed that p110δ is a crucial PI3K isoform involved in TCR-induced signaling.12,17,19 Here we show that both CD62L+ naive T cells and CD62L− effector/memory T cells are sensitive to inhibition with the p110δ-selective inhibitor IC. Indeed, effector/memory T cells appeared more sensitive to IC, suggesting that it might be possible to tune the level of p110δ inhibition in the clinic to preferentially affect pathogenic T cells. We also demonstrated that signals transduced by p110δ 24 hours or more after initial TCR stimulation can potently promote IFN-γ production, suggesting that p110δ inhibitors may be effective during an ongoing immune response. Chronic inactivation of p110δ in p110δD910A mice or acute p110δ inactivation with IC partially blocked CHS in mice during primary and secondary responses. These and previous studies18,32,33 suggest that p110δ inhibitors have promising therapeutic potential for the treatment of T-cell–mediated pathologies. However, no studies had directly addressed the role of p110δ in TCR activation of human T cells, and the possibility remained that additional PI3K isoforms might render p110δ redundant. This appears not to be the case, as IC inhibited anti-CD3 and anti-CD28 stimulated phosphorylation of Akt and reduced phosphorylation of Akt targets S6 and GSK3β at a concentration where it does not inhibit other PI3K isoforms. Blockade of TCR-induced p110δ signaling did not lead to generalized T-cell suppression, as CD69 up-regulation was unaffected except at the highest doses of inhibitor. Instead, p110δ inhibition had a selective effect on cytokine production, which could be reduced or completely blocked in effector/memory T cells. We had previously demonstrated in mice that naive T cells require p110δ activity for differentiation to T helper subsets19 ; here we extend this work by demonstrating that sustained p110δ activity controls cytokine production in differentiated T cells.
Reactivation of memory T cells is involved in disease maintenance in many inflammatory conditions, and blockade of their cytokine production may limit activation of accessory cells and tissue destruction. This is evidenced by elevated levels of TH2 cytokines in atopy and asthma and TH1 and TH17 cytokines in arthritis and chronic obstructive pulmonary disease.5,6 Modulation of cytokine responses can be efficacious in the clinic. The therapeutic benefit observed with corticosteroid treatment for asthma and TNF-α antagonists for arthritis and other autoimmune disorders, such as psoriasis, is thought to reflect suppression of the expression of inflammatory cytokines.6,7 However, severe asthmatics and patients with chronic obstructive pulmonary disease often resist corticosteroids, and many arthritis patients are refractory to TNF-α antagonists.5,6 Thus, additional strategies are needed to complement or supplement existing drugs. Here we show that p110δ inhibitors potently suppress TH2-associated cytokines produced by T cells from allergic patients. In addition, we show that, when mononuclear cells from sites of active inflammation in ReA patients were treated with IC, cytokine responses that directly drive disease were dampened. In rheumatoid arthritis, synovial fluid cells show elevated phosphorylation of Akt and other downstream molecules.50-52 It is therefore plausible that dysregulation of PI3K-dependent pathways may directly contribute to or drive inflammatory disease.
In conclusion, we demonstrate here that both mouse and human effector/memory cells use p110δ to promote cytokine production in T cells and that p110δ should therefore be considered as a target for therapeutic intervention of T cell–mediated pathology.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Anne Segonds-Pichon for help with statistical analysis and Bart Vanhaesebroeck and Fabien Garcon for critical review of manuscript.
This work was supported by the Biotechnology and Biological Sciences Research Council (K.O.) and the Norwegian Functional Genomics Program, the Research Council of Norway, the Norwegian Cancer Society, and Novo Nordisk Foundation (K.T.). D.R.S. was supported by a joint Collaborative Award in Science and Engineering from the Biotechnology and Biological Sciences Research Council and UCB Pharma.
Authorship
Contribution: D.R.S., E.B., K.M., V.Q.D., D.T.P., F.G., and P.B. performed experiments and analyzed data; K.M.T., B.T., K.T., P.B., and K.O. supervised the project and analyzed data; J.C. and J.S.H.G. provided essential materials and advice; D.R.S. wrote the paper with help from E.B. and K.O.; and all authors read and commented on a draft of the manuscript and contributed intellectually to the project.
Conflict-of-interest disclosure: F.G., B.T., and P.B. are or have been employees of a company that develops PI3K inhibitors. The remaining authors declare no competing financial interests.
Correspondence: Klaus Okkenhaug, Babraham Institute, Babraham Research Campus, Cambridge, CB22 3AT, United Kingdom; e-mail: klaus.okkenhaug@bbsrc.ac.uk.
