

Abstract
Preferentially expressed antigen of melanoma (PRAME) is aberrantly expressed in hematologic malignancies and may be a useful target for immunotherapy in leukemia. To determine whether PRAME is naturally immunogenic, we studied CD8+ T-cell responses to 4 HLA-A*0201–restricted PRAME-derived epitopes (PRA100, PRA142, PRA300, PRA425) in HLA-A*0201-positive patients with acute lymphoblastic leukemia (ALL), acute myeloid leukemia (AML), chronic myeloid leukemia (CML), and healthy donors. CD8+ T cells recognizing PRAME peptides could be detected ex vivo in 4 of 10 ALL, 6 of 10 AML, 3 of 10 CML patients, and 3 of 10 donors by HLA-A2 tetramer analysis and flow cytometry for intracellular interferon-γ. The frequency of PRAME-specific CD8+ T cells was greater in patients with AML, CML, and ALL than healthy controls. All peptides were immunogenic in patients, while responses were only detected to PRA300 in donors. High PRAME expression in patient peripheral blood mononuclear cells was associated with responses to greater than or equal to 2 PRAME epitopes compared with low PRAME expression levels (4/7 vs 0/23, P = .001), suggesting a PRAME-driven T-cell response. PRAME-specific T cells were readily expanded in short-term cultures in donors and patients. These results provide evidence for spontaneous T cell reactivity against multiple epitopes of PRAME in ALL, AML, and CML. The potential for developing PRAME as a target for immunotherapy in leukemia deserves further exploration.
Introduction
Preferentially expressed antigen of melanoma (PRAME) was first isolated as a human melanoma antigen by cDNA expression cloning using melanoma-reactive cytotoxic T cells (CTL).1 PRAME is a tumor antigen of particular interest because it is widely expressed by lymphoid and myeloid malignancies and solid tumors.2-4 PRAME appears to be instrumental in cancer progression, binding to retinoic-acid receptor-α, and inhibiting retinoic-acid-induced differentiation, growth arrest, and apoptosis.5 CTLs have been generated against 4 HLA-A*0201–restricted epitopes of PRAME, VLDGLDVLL (PRA100), SLYSFPEPEA (PRA142), ALYVDSLFFL (PRA300), and SLLQHLIGL (PRA425) in vitro.6-8 In addition, recent work demonstrated the presence of PRA100 and PRA300-specific T-cell responses in patients with acute myeloid leukemia (AML).9 Furthermore, vaccination with dendritic cells (DCs) generated from autologous leukemic blasts in patients with AML revealed a significant increase of CD8+ T cells specifically recognizing the PRA300 peptide, suggesting that this epitope is naturally processed and presented by AML cells.10 Similarly in patients with chronic myeloid leukemia (CML), CD8+ T cells against PRA300 could be successfully elicited in vitro after 2 rounds of stimulation with DCs transfected with Ph+ CML-RNA.11 PRAME is also overexpressed on the surface of acute lymphoblastic leukemia (ALL) cells;12,13 however, its role as a target antigen in ALL remains unexplored.
In this study, we determined the natural immunogenicity of PRA100, PRA142, PRA300, and PRA425 in HLA-A*0201–positive patients with ALL, AML, CML, and healthy donors. CD8+ T-cell responses against PRAME were detected directly ex vivo in patients with AML, CML, and ALL and at low frequencies in healthy donors, suggesting a possible role for PRAME as a target for immunotherapy in leukemia patients.
Methods
Patients and healthy controls
HLA-A*0201–positive patients with AML (n = 10), ALL (n = 10), CML (n = 10), solid tumors including esophageal carcinoma, colon adenocarcinoma, and sarcoma (n = 5), and HLA-A*0201-positive healthy donors (n = 12) were studied. Cells from patients and human leukocyte antigen (HLA)–identical donors were obtained from leukapheresis products (LP) before stem cell transplant (SCT). Patient 1 was in relapse before second allo-SCT. All patients and donors gave written informed consent on treatment protocols approved by the National Institutes of Health (NIH) Institutional Review Board in accordance with the Declaration of Helsinki. Peripheral blood mononuclear cells (PBMCs) were separated using Ficoll-Hypaque density gradient centrifugation (Organon-Teknika, Durham, NC) and cryopreserved in RPMI-1640 complete medium (CM; Life Technologies, Gaithersburg, MD) supplemented with 20% heat-inactivated fetal calf serum (FCS) and 10% dimethyl-sulfoxide according to standard protocols. Cells were thawed for experiments, washed, and suspended in RPMI-CM plus 10% pooled human AB serum (Sigma-Aldrich, St Louis, MO). High-resolution HLA class I genotyping was performed by sequence-specific polymerase chain reaction (PCR) using genomic DNA (HLA-Laboratory, Department of Transfusion Medicine, NIH, Bethesda, MD). The presence of immunoglobulin-G (IgG) and IgM cytomegalovirus (CMV) antibodies in the samples was analyzed by passive latex agglutination (CMVSCAN kit; Becton Dickinson Microbiology System, Cockeysville, MD).
Peptide synthesis
Peptides were prepared by Biosynthesis (Lewisville, TX) to a minimum purity of 95%. The identity of each of the peptides was confirmed by mass spectral analysis. The following peptides were tested: VLDGLDVLL (PRA100), SLYSFPEPEA (PRA142), ALYVDSLFFL (PRA300), SLLQHLIGL (PRA425),6 CMVpp65495 (NLVPMVATV),14 gp100209-217(210M) (IMDQVPFSV)15 and CAP1-6D (YLSGADLNL).16
Peptide-HLA class I tetrameric complexes and immunophenotyping
Unmanipulated PBMCs obtained from patients and donors were stained with allophycocyanin (APC)–conjugated PRA300/HLA-A*0201, CMVpp65495/HLA-A*0201 (positive control), HLA-A2 Null (negative control; all from Beckman-Coulter, Fullerton, CA). Sample staining was performed using 106 PBMCs in 50 μL 1% FCS/phosphate-buffered saline (PBS). Tetramers were added for 20 to 30 minutes at 37°C. Cells were washed once in 1% FCS/PBS and then stained with a titrated panel of directly conjugated antibodies to CD3, CD8, CD27, and CD45RO (Beckman-Coulter, Miami, FL). Fluorescein-isothiocyanate (FITC), phycoerythrin (PE), peridinin chlorophyll protein (PerCP), and PE-Cy7 were used as fluorophores. The lymphocytes were then washed in 1% bovine serum albumin (BSA)/PBS, and resuspended in 1% paraformaldehyde in PBS. A minimum of 0.5 × 106 gated cells were acquired. Flow cytometry was performed on an LSRII flow cytometer using FacsDiva software (both from BD Biosciences, San Jose, CA).
Flow cytometric detection of functional antigen-specific CD8+ T cells
PBMCs were adjusted to 106 cells/mL and costimulatory antibodies (αCD28 and 49d, 1 μg/mL; BD Biosciences), monensin (Golgistop, 0.7μL/mL; BD Biosciences), brefeldin-A (10 μg/mL; Sigma-Aldrich), and αCD107a-FITC (BD, San Diego, CA) added. The cells were loaded with test peptides, PRA100, PRA142, PRA300, and PRA425, (0.1 and 10 μM). An unstimulated tube and 2 positive controls (Staphylococcus enterotoxin-B or SEB, 1μg/mL; Sigma-Aldrich) and CMVpp65495, were included in each assay. Cells were incubated for 5 hours at 37°C. Cells were then washed and stained with an anti-CD3 PerCP-conjugated antibody and anti-CD8 FITC- or APC-conjugated antibody, fixed/permeablized, and then stained with an anti–interferon-γ (anti–IFN-γ), anti–interleukin-4 (anti–IL-4), anti–IL-2, anti–IL-10 FITC, or APC-conjugate (all BD/Pharmingen, San Diego, CA).
Short-term expansion of PRAME-specific CD8+ T cells in vitro
Briefly, PBMCs were thawed and suspended in AIM-V (Invitrogen, Carlsbad, CA) and 5% human AB serum (Valley Biomedical, Winchester, VA) hereafter referred to as AIM/HS, with or without IL-12 (2 ng/mL; Peprotech, Rocky Hill, NJ). Cells were plated in 96-well round bottom microtiter plates (100 000-200 000/well) and loaded with test peptides (0.1 and 10 μg/mL) or medium alone (no peptide-negative control). Test peptides included PRA100, PRA142, PRA300, and PRA425, and control peptides gp100209-217(210M) and CAP1-6D (irrelevant HLA-A*0201–binding peptide-negative controls), and CMVpp65495 (positive control for CMV-responsive patients/donors) were added directly to each well. The microtiter plates were incubated at 37°C with 5% CO2. On day 4, 100 μL AIM/HS containing recombinant human IL-2 (10 IU/mL; Peprotech) were added. Cells were harvested on day 7 for enzyme-linked immunosorbent spot (ELISPOT) analysis.
IFN-γ ELISPOT assay
The frequency of antigen-specific T cells in freshly thawed PBMCs and in PBMCs expanded for 7 days in vitro was assessed using a modification of an assay described earlier.17,18 Briefly, anti–IFN-γ–coated ELISPOT plates were prepared by sequentially treating ImmunoSpot MultiScreen HTS IP 96-well plates (Millipore, Watertown, MA) with 70% methanol in water for 1 to 2 minutes at room temperature followed by IFN-γ capture antibody (10 μg/mL) overnight at 4°C, and BSA (10 mg/mL in PBS) for 2 hours at 37°C. To measure PRAME-specific T-cell responses directly, 100 000 to 200 000 viable, freshly thawed PBMCs were incubated for 18 hours in anti–IFN-γ–coated ELISPOT wells with transporter associated with antigen processing (TAP)–defective T2 cells (50 000 cell/well) loaded with each of the following peptides: individual PRAME peptides (0.1 or 10 μg/mL), control peptides, gp100 or CAP-1-6D (irrelevant HLA-A*0201–binding peptide-negative controls), CMVpp65495 (positive control for CMV-responsive patients/donors), or not loaded with peptide. T2 cells were incubated with or without test peptide for 2 hours and then washed in standard media to remove excess peptide before use.
In 7-day expansion assays, antigen-specific T-cell response was enhanced by supplementing culture media with IL-12 (2 ng/mL). For each experiment, negative control wells containing responder cells and unpulsed autologous PBMCs and positive-control wells containing responder cells incubated with either CMVpp65495 peptide or phytohemagglutinin (1:100 dilution of commercially available reagent; Invitrogen) were included. On day 7, cells were stimulated with T2 cells loaded with or without test peptide, and ELISPOT responses were measured in 4-6 replicate wells. Spots were counted using an automated ELISPOT reader (AID, Strassberg, Germany) and the ELISPOT 3.1 SR software.
Equation for calculating SEM
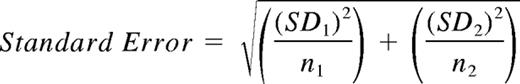
Where SD1 and SD2 represent the standard deviations for the number of spots produced by a specific peptide and by control antigen-presenting cells without peptide, and n1 and n2 are the number of replicate values for the number of spots generated.
Generation and expansion of antigen-specific CTLs
PBMCs were thawed and cultured in 96-, 48-, or 24-well plates in RPMI/10% normal AB serum (NABS) and IL-15 (25 ng/mL; PeproTech). Peptides (PRA100, PRA142, PRA300, PRA425) and CMVpp65495 (positive control) were added directly to the culture at 1 μg/mL. Recombinant human IL-2 (100 U/mL; Chiron, Emeryville, CA) was added on day 3, and cultures were restimulated on days 7, 14, and 21 with irradiated (25 Gy) peptide-loaded autologous PBMCs. IL-2 and IL-15 were added weekly. Intracellular cytokine assay was used to identify T-cell expansion in peptide-stimulated cultures.
Measurement of PRAME by real-time quantitative RT-PCR
RNA was isolated from a minimum of 106 PBMCs using RNeasy mini-kits (QIAGEN, Valencia, CA). cDNA was synthesized using the Advantage RT-for-PCR kit (Clontech, Mountain View, CA). ABL expression was used as the endogenous cDNA quantity control for all samples19 ; its expression was measured using 300 nM primers and 200 nM probe.20 Expression of PRAME was measured using TaqMan Assays-on-demand probe-and-primer reagents (Applied Biosystems, Foster City, CA) for PRAME, Hs00196132_m1, and expressed as a ratio of PRAME/ABL. All reactions by quantitative reverse-transcription (RT)–PCR using the ABI Prism 7900 sequence detection system (Applied Biosystems) were performed in triplicate in 10 μL volume using standard conditions with 40 cycles of amplification.
Statistical analysis
The Mann-Whitney test was used to determine whether there was a statistically significant difference in IFN-γ production in response to test peptides between patients with CML, AML, and ALL and healthy controls. The relationship between the number of peptides recognized and PRAME expression level was compared using the χ2 test. Statistical significance was achieved when P values were less than .05.
Results
Ex vivo detection of CD8+ T-cell responses directed against 4 HLA-A*0201–restricted PRAME epitopes
Samples from subjects in the cohort were studied for the presence of responses to PRA100, PRA142, PRA300, and PRA425. All AML and ALL patients had received prior chemotherapy. The clinical profiles of these donors and patients are presented in Table 1. We analyzed CD8+ T-cell responses to PRAME peptides by intracellular interferon-γ (IC-IFN–γ) staining. The data are summarized in Figure 1 and Table 1. A response was considered positive if the percentage of peptide-specific IFN-γ–producing CD8+ T cells was 2-fold or higher compared with the percentage of IFN-γ–producing CD8+ T cells in the absence of peptide and if there was a minimum of 0.05% peptide-specific IFN-γ–producing CD8+ T cells in 106 PBMCs (after subtracting the percentage of IFN-γ–producing CD8+ T cells in the absence of peptide). Significant peptide-specific IC-IFN–γ production to one or more peptides was observed in 6 of 10 patients with AML, 3 of 10 patients with CML, 5 of 10 with ALL, and 3 of 10 healthy donors. For donors 41 and 42, insufficient material was available to perform the analysis. There was no statistically significant difference in the frequency of PRAME T-cell responders in these groups. Analysis of the PRAME-responding population revealed that, in healthy donors, the PRAME-specific CD8+ T-cell response was limited to a single peptide, PRA300, in 3 of 3 cases. In contrast, 7 of 13 PRAME-responders with leukemia (2/6 AML, 3/3 CML, and 2/4 ALL) had detectable responses to 2 or more PRAME-derived peptides, suggesting that the presence of leukemia cells induces T-cell responses to several different PRAME epitopes.
Patient characteristics
| Diagnosis/patient no. | Disease status atanalysis | Percent blasts/cytogeneticsat analysis | PRAME/ABL (RT-PCR) | IFN-γ+ CD8+ T cells (%) | ||||
|---|---|---|---|---|---|---|---|---|
| PRA100 | PRA142 | PRA300 | PRA425 | |||||
| AML | ||||||||
| 1 | Ref | 5-10/Normal | 1.10035 | Neg | 0.06 | 0.11 | Neg | |
| 2 | Ref | 5-10/Normal | 0.00000 | Neg | Neg | Neg | Neg | |
| 3 | CR1 | < 5/Normal | 0.01235 | 0.14 | Neg | 0.11 | 0.06 | |
| 4 | CR1 | < 5/Normal | 0.00000 | Neg | Neg | 0.08 | Neg | |
| 5 | CR1 | < 5/Normal | 0.00140 | Neg | Neg | 0.10 | Neg | |
| 6 | CR2 | < 5/Normal | 0.00004 | Neg | Neg | Neg | Neg | |
| 7 | Ref | 11/Complex | 0.00000 | Neg | Neg | Neg | Neg | |
| 8 | CR1 | < 5/Normal | 0.00000 | Neg | Neg | Neg | Neg | |
| 9 | CR1 | < 5/Normal | 0.00015 | Neg | 0.25 | Neg | Neg | |
| 10 | Ref | < 5/Normal | 0.00000 | Neg | Neg | 0.06 | Neg | |
| Response | 6 of 10 | 1 | 2 | 5 | 1 | |||
| CML | ||||||||
| 11 | CML-AP | 8/Ph+ 100 | 0.00008 | Neg | Neg | Neg | Neg | |
| 12 | CML-CP | 5/Ph+ 100 | 0.00005 | Neg | Neg | Neg | Neg | |
| 13 | CML-CP | 5/Ph+ 95 | 0.00038 | Neg | Neg | Neg | Neg | |
| 14 | CML-BC | 22/Ph+ 100 | 0.00012 | Neg | Neg | Neg | Neg | |
| 15 | CML-CP | 3/Ph+ 100 | 0.00000 | 0.13 | 0.1 | Neg | Neg | |
| 16 | CML-CP | 4/Ph+ 100 | 0.00027 | Neg | Neg | Neg | Neg | |
| 17 | CML-AP | 13/Ph+ 100 | 0.00007 | 0.12 | Neg | 0.24 | 0.09 | |
| 18 | CML-CP | 5/Ph+ 100 | 0.00019 | Neg | Neg | Neg | Neg | |
| 19 | CML-AP | > 10/Ph+ 100 | 0.36629 | Neg | 0.09 | 0.15 | Neg | |
| 20 | CML-CP | < 10/Ph+ 100 | 0.00000 | Neg | Neg | Neg | Neg | |
| Response | 3 of 10 | 2 | 2 | 2 | 1 | |||
| ALL | ||||||||
| 21 | CR2 | < 5/Normal | 0.00000 | Neg | Neg | Neg | 0.11 | |
| 22 | Ph+ ALL- CR2 | < 5/Ph+ | 0.00000 | 0.29 | Neg | Neg | Neg | |
| 23 | Ref | 10/Normal | 0.00122 | 0.19 | Neg | 0.10 | Neg | |
| 24 | CR2 | < 5/Normal | 0.00000 | Neg | Neg | Neg | Neg | |
| 25 | CR2 | < 5/Normal | 0.00000 | Neg | 0.2 | 0.2 | Neg | |
| 26 | CR2 | < 5/Normal | 0.00016 | Neg | Neg | Neg | Neg | |
| 27 | CR2 | < 5/Normal | 0.00000 | Neg | Neg | 0.13 | Neg | |
| 28 | CR2 | < 5/Normal | 0.00000 | Neg | Neg | Neg | Neg | |
| 29 | Ref | 8/Normal | 0.00000 | Neg | Neg | Neg | Neg | |
| 30 | CR3 | < 5/Normal | 0.00000 | Neg | Neg | Neg | Neg | |
| Response | 5 of 10 | 2 | 1 | 3 | 1 | |||
| Donor | ||||||||
| 31 | 0.00000 | Neg | Neg | Neg | Neg | |||
| 32 | 0.00000 | Neg | Neg | Neg | Neg | |||
| 33 | 0.00000 | Neg | Neg | Neg | Neg | |||
| 34 | 0.00035 | Neg | Neg | 0.12 | Neg | |||
| 35 | 0.00000 | Neg | Neg | Neg | Neg | |||
| 36 | 0.00000 | Neg | Neg | Neg | Neg | |||
| 37 | 0.00000 | Neg | Neg | Neg | Neg | |||
| 38 | 0.00119 | Neg | Neg | 0.09 | Neg | |||
| 39 | 0.00000 | Neg | Neg | Neg | Neg | |||
| 40 | 0.00142 | Neg | Neg | 0.06 | Neg | |||
| Response | 3 of 10 | 0 | 0 | 3 | 0 | |||
| Solid tumor | ||||||||
| ST1 | Sarcoma | 0.00064 | Neg | Neg | Neg | Neg | ||
| ST2 | Colon carcinoma | 0.00000 | Neg | Neg | Neg | Neg | ||
| ST3 | Sarcoma | 0.00003 | Neg | Neg | Neg | Neg | ||
| ST4 | Colon carcinoma | 0.00000 | Neg | Neg | Neg | Neg | ||
| ST5 | Esophageal carcinoma | N/A | Neg | Neg | Neg | Neg | ||
| Response | 0 of 5 | 0 | 0 | 0 | 0 | |||
| Diagnosis/patient no. | Disease status atanalysis | Percent blasts/cytogeneticsat analysis | PRAME/ABL (RT-PCR) | IFN-γ+ CD8+ T cells (%) | ||||
|---|---|---|---|---|---|---|---|---|
| PRA100 | PRA142 | PRA300 | PRA425 | |||||
| AML | ||||||||
| 1 | Ref | 5-10/Normal | 1.10035 | Neg | 0.06 | 0.11 | Neg | |
| 2 | Ref | 5-10/Normal | 0.00000 | Neg | Neg | Neg | Neg | |
| 3 | CR1 | < 5/Normal | 0.01235 | 0.14 | Neg | 0.11 | 0.06 | |
| 4 | CR1 | < 5/Normal | 0.00000 | Neg | Neg | 0.08 | Neg | |
| 5 | CR1 | < 5/Normal | 0.00140 | Neg | Neg | 0.10 | Neg | |
| 6 | CR2 | < 5/Normal | 0.00004 | Neg | Neg | Neg | Neg | |
| 7 | Ref | 11/Complex | 0.00000 | Neg | Neg | Neg | Neg | |
| 8 | CR1 | < 5/Normal | 0.00000 | Neg | Neg | Neg | Neg | |
| 9 | CR1 | < 5/Normal | 0.00015 | Neg | 0.25 | Neg | Neg | |
| 10 | Ref | < 5/Normal | 0.00000 | Neg | Neg | 0.06 | Neg | |
| Response | 6 of 10 | 1 | 2 | 5 | 1 | |||
| CML | ||||||||
| 11 | CML-AP | 8/Ph+ 100 | 0.00008 | Neg | Neg | Neg | Neg | |
| 12 | CML-CP | 5/Ph+ 100 | 0.00005 | Neg | Neg | Neg | Neg | |
| 13 | CML-CP | 5/Ph+ 95 | 0.00038 | Neg | Neg | Neg | Neg | |
| 14 | CML-BC | 22/Ph+ 100 | 0.00012 | Neg | Neg | Neg | Neg | |
| 15 | CML-CP | 3/Ph+ 100 | 0.00000 | 0.13 | 0.1 | Neg | Neg | |
| 16 | CML-CP | 4/Ph+ 100 | 0.00027 | Neg | Neg | Neg | Neg | |
| 17 | CML-AP | 13/Ph+ 100 | 0.00007 | 0.12 | Neg | 0.24 | 0.09 | |
| 18 | CML-CP | 5/Ph+ 100 | 0.00019 | Neg | Neg | Neg | Neg | |
| 19 | CML-AP | > 10/Ph+ 100 | 0.36629 | Neg | 0.09 | 0.15 | Neg | |
| 20 | CML-CP | < 10/Ph+ 100 | 0.00000 | Neg | Neg | Neg | Neg | |
| Response | 3 of 10 | 2 | 2 | 2 | 1 | |||
| ALL | ||||||||
| 21 | CR2 | < 5/Normal | 0.00000 | Neg | Neg | Neg | 0.11 | |
| 22 | Ph+ ALL- CR2 | < 5/Ph+ | 0.00000 | 0.29 | Neg | Neg | Neg | |
| 23 | Ref | 10/Normal | 0.00122 | 0.19 | Neg | 0.10 | Neg | |
| 24 | CR2 | < 5/Normal | 0.00000 | Neg | Neg | Neg | Neg | |
| 25 | CR2 | < 5/Normal | 0.00000 | Neg | 0.2 | 0.2 | Neg | |
| 26 | CR2 | < 5/Normal | 0.00016 | Neg | Neg | Neg | Neg | |
| 27 | CR2 | < 5/Normal | 0.00000 | Neg | Neg | 0.13 | Neg | |
| 28 | CR2 | < 5/Normal | 0.00000 | Neg | Neg | Neg | Neg | |
| 29 | Ref | 8/Normal | 0.00000 | Neg | Neg | Neg | Neg | |
| 30 | CR3 | < 5/Normal | 0.00000 | Neg | Neg | Neg | Neg | |
| Response | 5 of 10 | 2 | 1 | 3 | 1 | |||
| Donor | ||||||||
| 31 | 0.00000 | Neg | Neg | Neg | Neg | |||
| 32 | 0.00000 | Neg | Neg | Neg | Neg | |||
| 33 | 0.00000 | Neg | Neg | Neg | Neg | |||
| 34 | 0.00035 | Neg | Neg | 0.12 | Neg | |||
| 35 | 0.00000 | Neg | Neg | Neg | Neg | |||
| 36 | 0.00000 | Neg | Neg | Neg | Neg | |||
| 37 | 0.00000 | Neg | Neg | Neg | Neg | |||
| 38 | 0.00119 | Neg | Neg | 0.09 | Neg | |||
| 39 | 0.00000 | Neg | Neg | Neg | Neg | |||
| 40 | 0.00142 | Neg | Neg | 0.06 | Neg | |||
| Response | 3 of 10 | 0 | 0 | 3 | 0 | |||
| Solid tumor | ||||||||
| ST1 | Sarcoma | 0.00064 | Neg | Neg | Neg | Neg | ||
| ST2 | Colon carcinoma | 0.00000 | Neg | Neg | Neg | Neg | ||
| ST3 | Sarcoma | 0.00003 | Neg | Neg | Neg | Neg | ||
| ST4 | Colon carcinoma | 0.00000 | Neg | Neg | Neg | Neg | ||
| ST5 | Esophageal carcinoma | N/A | Neg | Neg | Neg | Neg | ||
| Response | 0 of 5 | 0 | 0 | 0 | 0 | |||
The figures in bold represent positive CD8+ T-cell responses to stimulation with PRAME peptide.
AML indicates acute myeloid leukemia; CR, complete remission; CML, chronic myeloid leukemia; CP, chronic phase; AP, accelerated phase; BC, blast crisis; ALL, acute lymphoblastic leukemia; Ph, Philadelphia; Ref, refractory; D, donor; and N/A, not applicable.

CD8+ T-cell responses defined by analysis of intracellular cytokine production. (A) AML patient 3. (B) CML patient 17. (C) ALL patient 23. (D) Healthy donor 34. PBMCs (106) were loaded with test peptides, and intracellular cytokine analysis for IFN-γ was performed as described in “Methods.” Plots are gated on CD3+ T cells. Numbers in the upper right quadrant represent the percentage of IFN-γ–producing CD8+ T cells.
CD8+ T-cell responses defined by analysis of intracellular cytokine production. (A) AML patient 3. (B) CML patient 17. (C) ALL patient 23. (D) Healthy donor 34. PBMCs (106) were loaded with test peptides, and intracellular cytokine analysis for IFN-γ was performed as described in “Methods.” Plots are gated on CD3+ T cells. Numbers in the upper right quadrant represent the percentage of IFN-γ–producing CD8+ T cells.
To further ascertain that the PRAME CD8+ T-cell response was indeed specific to the disease, we studied 5 patients with solid tumors after chemotherapy. These included 2 patients with sarcoma, 2 with colon adenocarcinoma, and one with esophageal adenocarcinoma (Table 1). All patients had normal peripheral blood counts at analysis. PRAME expression was detected in the peripheral blood of 2 patients with sarcoma. Overexpression of PRAME in sarcoma has been previously reported.21 None of the 5 patients with solid tumors mounted a CD8+ T-cell response to PRAME. The absence of a significant PRAME response in patients with solid tumor further supports the notion that the PRAME-specific CD8+ T-cell response is indeed appropriate for the presence of the leukemia antigen and not merely due to previous chemotherapy or concurrent disease. Given that 3 of 10 healthy donors have responses to PRAME, we would expect a comparable proportion of people with illnesses unrelated to leukemia, such as those with solid tumors, to respond in a similar way, but in our series this was not observed. A possible explanation may be that solid tumor patients are often more heavily pretreated with chemotherapy than leukemia patients, which may lead to a state of generalized immune-suppression. Such small PRAME-CTL responses, if originally present, may have been further suppressed to undetectable levels, although we could detect CD8+ T-cell responses against SEB and CMVpp65495 in CMV-seropositive patients with solid tumor (data not shown).
PRAME expression in patients with leukemia and healthy donors
PRAME expression was detected in 2 of 10 patients with ALL (median 0, range 0-0.00121 PRAME/ABL), 5 of 10 patients with AML (median 0.00002, range 0-1.10035 PRAME/ABL), 8 of 10 patients with CML (median 0.00010, range 0-0.36630 PRAME/ABL), and 3 of 10 healthy donors (median 0, range 0-0.00142; Table 1). To determine whether there is an association between PRAME gene expression and the extent of PRAME-specific CD8+ T-cell response, we compared the number of PRAME epitopes eliciting a CD8+ T-cell response, using an arbitrary threshold for PRAME expression based on levels detected in healthy donors, in patients with levels of greater than 0.001 PRAME/ABL compared with those with levels of less than 0.001 PRAME/ABL. Expression of greater than 0.001 PRAME/ABL was significantly associated with a multiepitopic PRAME-specific CD8+ T-cell response (P = .001). Four of 6 patients with ALL, AML, or CML with PRAME expression levels of greater than 0.001 PRAME/ABL had responses to 2 or more epitopes of PRAME, whereas only 2 of 22 patients with PRAME levels of less than 0.001 PRAME/ABL had responses to more than one peptide (Figure 2). CD8+ T-cell responses to PRA300 were only seen in healthy donors with detectable PRAME gene expression, further supporting the immunogenicity of PRAME in vivo.

Relationship between PRAME gene expression and the number of PRAME epitopes recognized by CD8+ T cells in patients with AML, CML, ALL, and donors. Bars represent median values.
Relationship between PRAME gene expression and the number of PRAME epitopes recognized by CD8+ T cells in patients with AML, CML, ALL, and donors. Bars represent median values.
Extent and avidity of CD8+ T-cell response to PRAME
The frequencies of PRAME-specific CD8+ T cells as measured by IC-IFN–γ production to peptide stimulation was greater in patients with myeloid leukemia (AML and CML) and ALL compared with healthy controls (Figure 3). In view of the small number of subjects, it was not possible to compare the response to each individual peptide, although the response to all 4 peptides was significantly greater in patients with myeloid leukemia compared with healthy donors (P = .01). Similarly, the response was significantly greater in ALL patients compared with healthy donors (P = .04). The response to all 4 peptides was not significantly different in patients with myeloid leukemia compared with patients with ALL (P = .37; Figure 3A).

CD8+ T-cell response to PRAME in patients and donors. (A) Cumulative CD8+ T-cell response to stimulation with all 4 PRAME peptides, PRA100, PRA142, PRA300, and PRA425, in patients with myeloid leukemia (AML and CML), patients with ALL, and healthy donors. CD8+ T-cell responses to stimulation with the HLA-A*0201–restricted peptides: (B) PRA100, (C) PRA142, (D) PRA300, and (E) PRA425 in patients with leukemia (AML, ALL, and CML) compared with healthy donors. Values represent frequencies of PRAME-specific CD8+ T cells. (F) High- and low-avidity CD8+ T-cell responses determined by sensitivity to peptide concentration. Stimulation of PBMCs with 0.1 and 10 μM of PRA100, PRA142, PRA300, and PRA425 determined high- and low-avidity responses, respectively. Results shown are the ratios of high- to low-avidity CD8+ T-cell responses, calculated for patients with PRAME greater than 0.001 (○) and for patients with PRAME less than 0.001 (●). Ratios were obtained by the following calculation: IFN-γ plus CD8+ T cell (%) with 0.1 μM peptide/IFN-γ plus CD8+ T cell (%) with 10 μM peptide. Bars represent the median high/low-avidity ratio for each peptide.
CD8+ T-cell response to PRAME in patients and donors. (A) Cumulative CD8+ T-cell response to stimulation with all 4 PRAME peptides, PRA100, PRA142, PRA300, and PRA425, in patients with myeloid leukemia (AML and CML), patients with ALL, and healthy donors. CD8+ T-cell responses to stimulation with the HLA-A*0201–restricted peptides: (B) PRA100, (C) PRA142, (D) PRA300, and (E) PRA425 in patients with leukemia (AML, ALL, and CML) compared with healthy donors. Values represent frequencies of PRAME-specific CD8+ T cells. (F) High- and low-avidity CD8+ T-cell responses determined by sensitivity to peptide concentration. Stimulation of PBMCs with 0.1 and 10 μM of PRA100, PRA142, PRA300, and PRA425 determined high- and low-avidity responses, respectively. Results shown are the ratios of high- to low-avidity CD8+ T-cell responses, calculated for patients with PRAME greater than 0.001 (○) and for patients with PRAME less than 0.001 (●). Ratios were obtained by the following calculation: IFN-γ plus CD8+ T cell (%) with 0.1 μM peptide/IFN-γ plus CD8+ T cell (%) with 10 μM peptide. Bars represent the median high/low-avidity ratio for each peptide.
All 4 peptides were immunogenic in patients with AML, CML, and ALL (Table 1 and Figure 3B-E). PRA100, PRA142, PRA300, and PRA425 were highly antigenic, each eliciting responses in 3 of 20, 4 of 20, 7 of 20, and 2 of 20 of patients with myeloid leukemia and 2 of 10, 1 of 10, 3 of 10, and 1 of 10 patients with ALL, respectively. In healthy donors however, responses could only be detected to PRA300 in 3 of 10 donors (Figure 3D). Peptides PRA100, PRA142, and PRA425 were highly specific for patients; none of the healthy donors exhibited responses to these peptides. The difference was statistically significant for PRA100 and PRA142 (P < .05), but did not reach statistical significance for PRA425 (P = .08; Figure 3B-E).
To determine the functional avidity of the PRAME T-cell response, the response of CD8+ T cells to stimulation with 2 concentrations of peptide (0.1 and 10 μM) was measured by IC-IFN–γ staining. High- and low-avidity T cells have been previously shown to possess different requirements for both peptide/major histocompatibility complex (MHC) density and CD8 interaction.22 We defined high-avidity CD8+ T cells as those capable of producing IFN-γ in response to a lower concentration of peptide (0.1 μM), while low-avidity CD8+ T cells were those that produced IFN-γ in response to a higher concentration of peptide (10 μM). These 2 peptide concentrations were chosen in this study because they were previously shown to stimulate and differentiate high- and low-avidity T-cell responses to other “self-antigens” such as PR1.23 We determined the ratio of high- to low-avidity CD8+ T-cell responses to each PRAME epitope by comparing the frequency of PRAME-specific CD8+ T cells to stimulation with low or higher peptide concentrations, respectively (Figure 3F). We had IC-IFN–γ data for both peptide dose in 37 of 40 subjects. For all peptides tested, both high- and low-avidity CD8+ T-cell responses could be detected (median 1.05, 0.90, 0.52, 0.40 high/low-avidity ratios for PRA100, PRA142, PRA300, and PRA425, respectively). When we compared patients with relatively high expression level of PRAME (> 0.001 PRAME/ABL) to those with low levels of PRAME expression (< 0.001 PRAME/ABL), we observed that in patients with (> 0.001 PRAME/ABL) low-avidity CD8+ T-cell responses to PRAME peptides were more prominent than high-avidity responses (Figure 3F, open circles), suggesting selective deletion of high-avidity T cells. In contrast, in some patients with low PRAME expression levels (< 0.001 PRAME/ABL), we could detect the presence of high-avidity CD8+ T-cell responses to PRAME. In view of the small sample size, it was not possible to compare the avidity of response to individual PRAME epitopes for each patient group and donors separately.
Functional characterization of PRAME-specific CD8+ T-cell response
To further assess the functionality of the PRAME-specific CD8+ T-cell response, 17 samples shown to have detectable PRAME-specific CD8+ T cells by IC-IFN–γ staining were analyzed by performing flow-cytometric analysis for IL-2, IL-4, and IL-10 production and CD107a mobilization (as a marker of cytotoxicity).24 After stimulation with the relevant PRAME peptide, there was no significant production of IL-2, IL-4, or IL-10, suggesting a Tc1 effector response. In 11 patients, the background staining with the CD107a antibody was too high to allow discrimination between stimulated and nonstimulated cells. In 6 patients (patients 3, 5, 9, 19, 22, 23) no significant CD107a mobilization was detected after stimulation with the relevant PRAME peptide. To exclude the possibility that the limited functionality of the CD8+ T-cell response in patients with leukemia may be due to a generalized immune-suppression phenomenon, we also tested the CD8+ T-cell response against CMVpp65495 and SEB. Significant IFN-γ production and CD107a mobilization was seen in response to CMVpp65495 (representative data from patient 9 are presented in Figure 4) and IFN-γ, IL-2, and CD107a T cells were detected in response to SEB (data not shown).

Characterization of PRAME-specific and CMV-specific CD8+ T-cell functionality in patients with leukemia. Shown are representative data of the PRAME- and CMV-specific CD8+ T-cell response from patient 9, after a 5-hour in vitro stimulation. See “Methods” for a detailed explanation of the procedure.
Characterization of PRAME-specific and CMV-specific CD8+ T-cell functionality in patients with leukemia. Shown are representative data of the PRAME- and CMV-specific CD8+ T-cell response from patient 9, after a 5-hour in vitro stimulation. See “Methods” for a detailed explanation of the procedure.
Phenotype of PRAME-specific CD8+ T cells
Sufficient material was available to perform phenotypic analysis of PRAME specific T cell responses in 2 patients only (patients 1 and 25). Antigen-specific CD8+ T cells identified by PRA300/HLA-A*0201 tetramers were analyzed in unmanipulated PBMCs for expression of CD45RO and CD27 to characterize naive, memory, effector, and late differentiated effector phenotype.25 The phenotypic analysis of patient 25 is illustrated in Figure 5. CMVpp65495/HLA-A*0201 plus CD8+ T cells were used as a positive control and HLA-A2 null as negative control. CMVpp65495/HLA-A*0201 plus CD8+ T cells exhibited a predominant phenotype of effector memory cells (CD45RO+CD27−) and late differentiated effector (CD45RO-CD27-) with a smaller population of central memory cells (CD45RO+CD27+). PRA300/HLA-A*0201 plus CD8+ T cells on the other hand were a mixture of effector and central memory phenotypes with a smaller population of late differentiated effector cells (Figure 5).

Phenotypic characterization of tetramer-positive CD3+CD8+ T cells. CMVpp65495/HLA-A*0201 CD8+ T cells were used as a positive control and HLA-A2 null as negative control. (A) Peptide/HLA-A2 tetramer analysis of PBMCs was performed by 6-color flow cytometry in 2 patients. (B) CD45RO and CD27 phenotype of CD3+ CD8+ T-cell gated tetramer-positive lymphocytes on samples from patient 25.
Phenotypic characterization of tetramer-positive CD3+CD8+ T cells. CMVpp65495/HLA-A*0201 CD8+ T cells were used as a positive control and HLA-A2 null as negative control. (A) Peptide/HLA-A2 tetramer analysis of PBMCs was performed by 6-color flow cytometry in 2 patients. (B) CD45RO and CD27 phenotype of CD3+ CD8+ T-cell gated tetramer-positive lymphocytes on samples from patient 25.
PRAME-specific CD8+ T cells expand in short-term cultures
To further define the functional capacity of PRAME-specific CD8+ T cells detected by cytokine secretion, cultured ELISPOT assays were performed after short-term expansion with peptide in 3 patients with AML, 5 patients with ALL, 3 patients with CML, and 4 healthy donors (Table 2; Figure S1A, available on the Blood website; see the Supplemental Materials link at the top of the online article). PBMCs from patients and donors were tested for their recognition of cognate peptide in an IFN-γ ELISPOT assay on day 1 and after 7 days in culture. A T-cell response was considered positive if there was a minimum of 20 peptide-specific spots in 106 PBMCs (after subtracting the number of spots in unstimulated PBMCs), if the number of spots in peptide-exposed PBMCs was 2-fold or more higher than the number of spots in unstimulated PBMCs and statistically significantly different from the control in comparison of the triplicates by Student t test. Cross-reactivity was excluded by testing responses against 2 HLA-A*0201–restricted irrelevant peptides, GP100 and CAP1-6D.
ELISPOT assay before and after short-term expansion
| Replicate | Prame 100 | Prame 142 | Peptide-specific ELISPOTs (spots/million PBMCs) | GP100/CAP-1–6D | CMV | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Prame 300 | Prame 425 | No peptide | |||||||||||||
| D1/D7 | D1 | D7 | D1 | D7 | D1 | D7 | D1 | D7 | D1 | D7 | D1 | D7 | |||
| AML | |||||||||||||||
| 1 | 6/6 | NEG | 21 ± 6 | NEG | 28 ± 7 | 131 ± 11 | 72 ± 15 | NEG | NEG | 3 | 3 | NEG | NEG | 330 ± 36 | 3500 ± 550 |
| 7 | 5/5 | NEG | NEG | NEG | NEG | NEG | NEG | NEG | NEG | 80 | 1 590 | NEG | NEG | NEG | NEG |
| 10 | 6/6 | NEG | 1410 ± 145 | 26 ± 6 | 230 ± 65 | 280 ± 60 | 550 ± 60 | NEG | 137 ± 45 | 2 | 50 | NEG | 660 ± 80 | 180 ± 52 | 7000 ± 165 |
| ALL | |||||||||||||||
| 21 | 5/5 | NEG | 1300 ± 65 | NEG | 2100 ± 35 | 2460 ± 40 | > 14 300 | NEG | 2500 ± 40 | 110 | 760 | NEG | 960 ± 90 | 1000 ± 110 | ND |
| 22 | 5/5 | NEG | NEG | NEG | 80 ± 23 | NEG | NEG | NEG | 150 ± 30 | 1 | 23 | NEG | 88 ± 31 | 25 ± 5 | ND |
| 23 | 5/5 | NEG | NEG | NEG | NEG | NEG | NEG | NEG | NEG | 2 | 12 | NEG | NEG | NEG | NEG |
| 24 | 5/5 | NEG | 1150 ± 200 | NEG | 3400 ± 360 | NEG | 3200 ± 290 | NEG | NEG | 24 | 320 | 36 ± 8 | NEG | 2950 ± 170 | ND |
| 25 | 5/5 | NEG | NEG | NEG | NEG | NEG | 660 ± 110 | NEG | 220 ± 60 | 18 | 95 | NEG | NEG | 540 ± 105 | ND |
| CML | |||||||||||||||
| 6/6 | NEG | NEG | NEG | NEG | NEG | 110 ± 20 | NEG | NEG | 70 | 65 | NEG | NEG | NEG | 282 ± 96 | |
| 6/6 | NEG | 28 ± 8 | NEG | NEG | NEG | NEG | NEG | NEG | 16 | 16 | NEG | NEG | 77 ± 26 | 205 ± 10 | |
| 6/6 | NEG | 2100 ± 300 | NEG | 2180 ± 180 | NEG | 550 ± 110 | NEG | 2220 ± 190 | 110 | 100 | NEG | 330 ± 100 | NEG | NEG | |
| Donors | |||||||||||||||
| 31 | 5/5 | NEG | NEG | NEG | NEG | 176 ± 27 | NEG | NEG | NEG | 150 | 135 | NEG | NEG | NEG | NEG |
| 35 | 5/5 | NEG | NEG | NEG | 980 ± 44 | 103 ± 40 | NEG | NEG | NEG | 77 | 130 | NEG | NEG | 303 ± 39 | > 3800 |
| 41 | 5/5 | NEG | NEG | NEG | NEG | NEG | NEG | NEG | NEG | 38 | 775 | NEG | 167 ± 94 | ND | |
| 42 | 4/5 | NEG | NEG | NEG | NEG | 99 ± 22 | 1630 ± 83 | NEG | NEG | 81 | 1126 | NEG | NEG | NEG | NEG |
| Replicate | Prame 100 | Prame 142 | Peptide-specific ELISPOTs (spots/million PBMCs) | GP100/CAP-1–6D | CMV | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Prame 300 | Prame 425 | No peptide | |||||||||||||
| D1/D7 | D1 | D7 | D1 | D7 | D1 | D7 | D1 | D7 | D1 | D7 | D1 | D7 | |||
| AML | |||||||||||||||
| 1 | 6/6 | NEG | 21 ± 6 | NEG | 28 ± 7 | 131 ± 11 | 72 ± 15 | NEG | NEG | 3 | 3 | NEG | NEG | 330 ± 36 | 3500 ± 550 |
| 7 | 5/5 | NEG | NEG | NEG | NEG | NEG | NEG | NEG | NEG | 80 | 1 590 | NEG | NEG | NEG | NEG |
| 10 | 6/6 | NEG | 1410 ± 145 | 26 ± 6 | 230 ± 65 | 280 ± 60 | 550 ± 60 | NEG | 137 ± 45 | 2 | 50 | NEG | 660 ± 80 | 180 ± 52 | 7000 ± 165 |
| ALL | |||||||||||||||
| 21 | 5/5 | NEG | 1300 ± 65 | NEG | 2100 ± 35 | 2460 ± 40 | > 14 300 | NEG | 2500 ± 40 | 110 | 760 | NEG | 960 ± 90 | 1000 ± 110 | ND |
| 22 | 5/5 | NEG | NEG | NEG | 80 ± 23 | NEG | NEG | NEG | 150 ± 30 | 1 | 23 | NEG | 88 ± 31 | 25 ± 5 | ND |
| 23 | 5/5 | NEG | NEG | NEG | NEG | NEG | NEG | NEG | NEG | 2 | 12 | NEG | NEG | NEG | NEG |
| 24 | 5/5 | NEG | 1150 ± 200 | NEG | 3400 ± 360 | NEG | 3200 ± 290 | NEG | NEG | 24 | 320 | 36 ± 8 | NEG | 2950 ± 170 | ND |
| 25 | 5/5 | NEG | NEG | NEG | NEG | NEG | 660 ± 110 | NEG | 220 ± 60 | 18 | 95 | NEG | NEG | 540 ± 105 | ND |
| CML | |||||||||||||||
| 6/6 | NEG | NEG | NEG | NEG | NEG | 110 ± 20 | NEG | NEG | 70 | 65 | NEG | NEG | NEG | 282 ± 96 | |
| 6/6 | NEG | 28 ± 8 | NEG | NEG | NEG | NEG | NEG | NEG | 16 | 16 | NEG | NEG | 77 ± 26 | 205 ± 10 | |
| 6/6 | NEG | 2100 ± 300 | NEG | 2180 ± 180 | NEG | 550 ± 110 | NEG | 2220 ± 190 | 110 | 100 | NEG | 330 ± 100 | NEG | NEG | |
| Donors | |||||||||||||||
| 31 | 5/5 | NEG | NEG | NEG | NEG | 176 ± 27 | NEG | NEG | NEG | 150 | 135 | NEG | NEG | NEG | NEG |
| 35 | 5/5 | NEG | NEG | NEG | 980 ± 44 | 103 ± 40 | NEG | NEG | NEG | 77 | 130 | NEG | NEG | 303 ± 39 | > 3800 |
| 41 | 5/5 | NEG | NEG | NEG | NEG | NEG | NEG | NEG | NEG | 38 | 775 | NEG | 167 ± 94 | ND | |
| 42 | 4/5 | NEG | NEG | NEG | NEG | 99 ± 22 | 1630 ± 83 | NEG | NEG | 81 | 1126 | NEG | NEG | NEG | NEG |
In all subjects, cross-reactivity was excluded by testing responses against the irrelevant peptides, GP100 or CAP1–6D. The absolute number of INF-γ spots from negative control wells (T2 cells with no peptide) is included. A T-cell response was defined as positive if there was a minimum of 20 peptide-specific spots in 106 PBMCs (after subtracting the number of spots in negative control wells), if the number of spots in peptide-exposed PBMCs was 2-fold or more higher than the number of spots the number of spots in negative control wells and statistically significantly different from the control by Student t test (P < .05). The mean plus or minus SEM values in positive wells represent spot numbers after subtracting the number of spots in negative control wells. The figures in bold represent positive CD8+ T-cell responses to stimulation with PRAME peptide.
D1 indicates assays performed prior to in vitro expansion; D7, assays performed after 7 days of in vitro expansion; and ND, not done.
On day 1, before in vitro expansion, a T-cell response could be detected to only one PRAME epitope, PRA300, in 3 of 4 healthy donors (donors 31, 35, and 42), with frequencies between 99 and 176 peptide-specific T cells in 106 PBMCs. After a 7-day in vitro expansion culture, PRA 300-specific T cells could be expanded in 1 of 3 donors (donor 42) with preexisiting PRA300-specific T-cell responses (1630 peptide-specific T cells in 106 PBMCs). Interestingly, after in vitro expansion, T-cell responses were observed to PRA142 (980 peptide-specific T cells in 106 PBMCs) in one healthy donor (donor 35), suggesting the presence of low frequencies of preexisting PRA142-specific T cells in this donor (Table 2).
Before in vitro expansion, T-cell responses to PRA300 or PRA142 could be detected in 2 of 3 patients with AML (patients 1 and 10), 0 of 3 patients with CML, and 1 of 5 patients with ALL (patient 21), with frequencies between 26 and 2457 peptide-specific T cells in 106 PBMCs (Table 2). After 7 days in vitro expansion culture, responses to PRA100, PRA142, PRA300, or PRA425 epitopes could be detected in 2 of 3 AML (patients 1 and 10), 3 of 3 CML (patients 15, 18, and 19), and 4 of 5 ALL patients (patients 21, 22, 24, and 25) with frequencies between 21 and 14 300 peptide-specific T cells in 106 PBMCs. These responses were directed against multiple epitopes in 2 of 2 AML, 1 of 3 CML, and 4 of 4 ALL PRAME responders, further supporting the immunogenicity of PRAME in leukemia. The cultured ELISPOT responses were of a much higher magnitude (mean ± SE 820 ± 60/106 PBMC) than the ex vivo responses (131 ± 27/106 PBMC), possibly reflecting their 1-week expansion period in vitro. Interestingly, in vitro culture expansion appeared more efficient in ALL than AML (21-550 vs 80-14 300; Table 2 and Figure S1A).
For 13 donors and patients in whom sufficient cells were available to perform all 3 assays (IC-IFN–γ, ex vivo ELISPOT, and cultured ELISPOT), we failed to find a significant correlation in the CD8+ T-cell response to PRAME (P = .65). The PBMCs were stimulated differently for each of the 3 T-cell assays, which may contribute to their lack of concordance, for example by preferentially promoting the activation of a different T-cell subset. Thus, with the IC-IFN–γ assay, a minimum of 106 PBMCs were cultured in complete RPMI media and stimulated directly with peptide for 5 hours. The cultured ELISPOT cells were cultured for 7 days in AIM/HS medium in a 96-well tissue culture plate, and recombinant human IL-2 was added. The ELISPOT assays using the ex vivo cells and cultured cells were performed by stimulating cells with T2 cells loaded with or without test peptide, and fewer cultured cells (105) were added per well because after culture, the reactive cells became too numerous to score visually.
We attempted to expand PRAME-specific T-cell lines as described in the Methods section. Long-term expansion of PRAME-specific CD8+ T cells in donors and patients with leukemia was not successful, although similar culture conditions allowed expansion of virus-specific T cells from the same patients. A representative data are presented in Figure S1B. This may reflect an inherent inability of PRAME-specific T cells to expand and could explain the failure of these natural immune responses to control leukemia growth.
Discussion
We used PRA300/HLA-A*0201 tetramer staining, intracellular cytokine assay and ex vivo and cultured ELISPOT analysis to study CD8+ T-cell responses to the cancer-testis antigen PRAME in patients with AML, ALL, and CML, and healthy donors. To broaden our understanding of T-cell responses to PRAME, we studied T-cell responses against 4 HLA-A*0201–restricted epitopes derived from this protein directly ex vivo and defined these responses in terms of their avidity and functional capacity. Our study shows the existence of naturally occurring memory CD8+ T cells directed against PRAME in patients with AML, CML, and ALL.
The detection of PRAME-specific T cells in leukemic patients suggests that the low level expression of PRAME in some healthy tissues does not induce irreversible tolerance against PRAME. Indeed detection of PRAME T-cell responses only in healthy donors with detectable peripheral blood PRAME gene expression further supports the natural immunogenicity of PRAME. These data are in keeping with earlier findings by our group and others that healthy people frequently have detectable numbers of circulating T cells that recognize leukemia-associated self-antigens (LAA) such as PR1 and WT1.26-30 CD8+ T-cell responses to LAA appear to be restricted and narrowly focused in healthy donors. In our study, we could only detect responses to PRA300, however after 1 week of in vitro expansion, responses were also detected to PRA142, suggesting the presence of low frequencies of preexisting PRA142-specific T cells. Similarly, using a 2-week in vitro expansion assay, Griffion et al could demonstrate the presence of PRA100 and PRA300-specific CD8+ T cells in 4 of 14 and 1 of 14 healthy donors, respectively,7 with PRA100-specific CD8+ T-cell precursor frequency in the order of 4 to 40 antigen-specific T cells in 106 CD8+ T cells (ie, 0.0004%-0.004%). Therefore, it appears that low frequencies of precursor T cells directed against PRAME epitopes exist in healthy donors, and their detection is often limited by the sensitivity of assays used. The demonstration of higher frequencies of CD8+ T cells recognizing PRAME epitopes in patients with AML, CML, and ALL compared with healthy donors suggests that patients with myeloid and ALL are naturally primed against these 4 epitopes. These results are in keeping with previous studies demonstrating the presence of CD8+ T-cell responses to PRA300 and PRA100 in patients with AML9 and to PRA300 in CML.11 To address the question of whether the responses seen in leukemia patients are specific and not merely a nonspecific response to prior chemotherapy, we studied 5 patients with solid tumors. None of the 5 patients with solid tumors had detectable T-cell responses to PRAME. These data suggest that the PRAME-specific CD8+ T-cell response is indeed appropriate for the presence of the leukemia antigen.
The IC-IFN–γ, ex vivo, and cultured ELISPOT assays gave differing reactivity patterns for all patients and donors tested, and no concordance was found among responses to the 3 assays. Similar observations of unique T-cell specificities to viral or tumor-associated antigens according to the assays used has been reported by several investigators.31-33 The reasons for such unique specificities in our study may partly reflect the different culture conditions used for the 3 assays, which, in turn, could have favored the expansion of different T-cell subsets. The medium for the ex vivo IC-IFN–γ (complete RPMI) was different from the ex vivo and cultured ELISPOT (AIM/HS), and IL-2 was added to the cultured ELISPOT cells. T2 cells pulsed with the relevant PRAME peptides were used to stimulate T cells in the ELISPOT assay on days 0 and 7, whereas in the IC-IFN–γ assay peptides were added directly to the cells, and costimulation using anti-CD28/CD49d was used. Importantly, the 3 assays are designed to detect different T-cell subsets. Immediate effector T-cell responses can be measured in an ex vivo 18-hour IFN-γ ELISPOT or 5-hour IC-IFN–γ assays and are thought to comprise mainly effector-memory T cells that circulate shortly after antigenic priming or recall.34 Resting memory T cells require antigenic restimulation, and we used the cultured IFN-γ ELISPOT, which reflects a potential IFN-γ–secreting T-cell capacity within PBMCs to measure such memory T cells.35 Therefore it appears that no single assay is as yet suitable for detecting the full breadth and functionality of the antigen-specific T-cell response, and the use of multiple assays as described here is likely to give the most information on immune responses to PRAME.
PRAME gene expression was detected in the peripheral blood of 5 of 10 patients with AML, 8 of 10 with CML, 2 of 10 with ALL, and 3 of 10 healthy donors. Low level detection of PRAME on normal CD34+ cells in healthy donors has been reported previously,13,36 although PRAME appears to be an adequate marker of minimal residual disease (MRD). Work published by several groups examining follow-up samples from patients with leukemia undergoing chemotherapy or allogeneic SCT demonstrated a strong correlation between PRAME expression and unique leukemia markers (AML1/ETO, PML-RARα, BCR-ABL).13,37,38 In all patients achieving complete remission after treatment, levels of PRAME decreased to levels comparable to healthy controls indicating that the expression of PRAME is not up-regulated in regenerating bone marrow or blood. In keeping with Griffioen et al,7 we found lower levels of PRAME expression in adults with ALL than previously reported.13,39 This may be because our patients had already received chemotherapy and most had achieved clinical remission at the time of study. Unfortunately, samples at diagnosis were not available for analysis. Griffioen et al failed to show recognition and killing of ALL cell lines by PRA100/HLA-A2 tetramer+ T-cell clones and suggested that PRAME expression in ALL is below the recognition threshold of their PRA100/HLA-A2 tetramer+ T-cell clones.7 However, in our study, the presence of high frequencies of CD8+ T cells against all 4 PRAME epitopes supports an immunogenic role for PRAME in ALL.
Our finding that patients with PRAME expression levels greater than 0.001 were significantly more likely to have responses to multiple PRAME epitopes compared with those with PRAME levels less than 0.001 may reflect the phenomenon of epitope spreading, thought to play a crucial role in several autoimmune disease models.40,41 It is noteworthy that many of the recently isolated tumor antigens, including PRAME, are nonmutated self-antigens inappropriately expressed or overexpressed in the tumor. We have shown previously a similar phenomenon with WT1, where CD8+ T cells directed against multiple epitopes of WT1 were detected in patients with myeloid leukemia.27 Effective cancer immunotherapy can thus be considered a “controlled” induction of autoimmunity, and some of the salient features of autoimmunity, including epitope spreading, are being recognized in the field of tumor immunology.42 Indirect evidence for the existence of epitope spreading in leukemia comes from work by our group and others, demonstrating that the majority of patients with myeloid leukemia have detectable immune responses to multiple leukemia antigens (eg, PR1, WT1, and BCR-ABL).26,29,30
It might be anticipated that the emergence of a leukemia expressing large amounts of PRAME should result in the extinction of PRAME-specific T cells and a narrowing of the T-cell response to PRAME. Indeed it has been clearly shown that in patients with leukemia, CTLs with the highest avidity for LAA, such as PR1, may be deleted.23 Similarly, recent work by Quintarelli et al demonstrated that CTL lines generated from CML patients were characterized by a lower avidity for PRAME-derived peptides than CTLs from normal donors.8 We found that in patients with relatively high expression of PRAME, despite an increase in the frequency of PRAME-specific CD8+ T cells and number of epitopes recognized, the responses were predominantly low-avidity, suggesting the selective deletion of high-avidity PRAME-specific CD8+ T cells. In contrast, high-avidity PRAME-specific T-cell responses were detected in some leukemia patients with low or undetectable PRAME expression. This may suggest the elimination of residual PRAME-positive leukemia cells by these CTLs. We have previously shown an inverse correlation of WT1 expression with the presence of WT1-specific CTLs associated with a graft-versus-leukemia (GVL) effect in leukemic patients after SCT.43 Similarly, our group and others have shown that in CML patients, a lower expression of PR3 or ELA2 was associated with PR1-CTL responses.44,45 Further longitudinal studies will be necessary to determine the relationship between leukemia and the T-cell response to PRAME and whether a reduction in the leukemia load and a state of minimal residual disease (MRD) will have an impact on the avidity of the PRAME-specific CD8+ T-cell response. Taken together, these data suggest that selective deletion of high-avidity LAA-specific CTLs in patients with leukemia may be a general immune evasion strategy.
The finding that in patients with leukemia, PRAME-specific CD8+ T cells were less efficient at the single cell level to mobilize CD107a than their CMV-specific counterparts demonstrates that the low cytotoxic activity is an expression of functional impairment that appears to be selective for PRAME-specific CD8+ T cells. Such selective functional unresponsiveness of antigen-specific CD8+ T cells has been reported in chronic viral infections including HIV and hepatitis C infection46,47 as well as in metastatic melanoma48 and fits with a model of exhaustion. However, the finding that PRAME-specific CD8+ T cells could be readily expanded in the presence of cytokines in short-term cultures in vitro to produce IFN-γ, suggests that it may be possible to improve the functional capacity of PRAME-specific T cells for therapeutic purposes.
The rapid expansion of PRAME-specific T cells in most patients and donors alike suggests the expansion was derived from preexisting memory CD8+ T cells. Our phenotype data in 2 patients with leukemia supports a memory origin for PRA300-specific T cells. These data are partly supported by previous work where variable numbers of memory PRA100/HLA-A2-tetramer+ CD8+ T cells were found in 2 healthy subjects and 1 melanoma patient.7 However, whereas PRA100/HLA-A2 tetramer+ CD8+ T cells in the majority of patients with melanoma had a naive phenotype, in our study, PRA-300/HLA-A2 tetramer+ CD8+ T cells analyzed in the 2 leukemia patients displayed a mixture of effector and memory phenotype. It may be that, in melanoma, T cells have not been activated in vivo by PRAME-expressing tumor cells, possibly due to lack of costimulatory molecules, defective DC function, or production of immunosuppressive factors, whereas PRAME-expressing leukemia cells are efficient at inducing an effective immune response. The presence of memory and effector T cells against several overexpressed leukemia-associated self-antigens, including PR1, WT1, surviving, and RHAMM, in patients with leukemia26-30,43,49,50 further supports the immunogenicity of leukemia cells. Moreover, CD8+ T cells with a memory phenotype against another cancer-testis antigen, MAGE, were recently reported in patients with myeloma.51
We were unable to expand PRAME-specific CD8+ T cells in long-term cultures in vitro, in contrast to virus-specific T cells from the same patients that expanded. This may reflect an inherent proliferative limitation of leukemia-associated CD8+ T cells as reported previously.43,52 Indeed Quintarelli et al succeeded in expanding CTLs from healthy donors and CML patients only in the presence of potent professional or artificial antigen presenting cells and cytokines.8 They demonstrated that PRAME-specific CD8+ T cells in healthy donors reside in the CD45RA+ T-cell compartment; however a similar analysis was not performed in patients with CML. We were unable to perform phenotypic analysis of PRA300-specific CD8+ T cells in healthy donors due to insufficient material. The failure to expand PRAME-specific T cells in vitro suggests that these PRAME-specific CD8+ T cells are anergized. Whether the detected frequencies of PRAME-reactive T cells could be sufficient to control residual disease cannot be ascertained from this study, and further longitudinal studies will be necessary to determine whether these cells have any impact on disease progression.
Although we were unable to obtain direct evidence that the PRAME-specific CD8+ T cells identified in these patients can exert lytic functions, their ability to proliferate in short-term cultures and to produce IFN-γ in vitro suggests that appropriate stimulation and/or addition of growth factors may overcome this ineffectiveness in vivo. All 4 PRAME epitopes tested were found to be immunogenic in patients with leukemia. Vaccination of leukemic patients with a cocktail of PRAME-peptides may therefore represent a simple and hence attractive approach. Our group and others have shown the feasibility of vaccination with peptides derived from other leukemia-associated antigens including PR1 and WT1.53-56 Furthermore, we recently demonstrated that PRAME is expressed in primitive CD34+ leukemic progenitors in CML,57 which can act as a reservoir for leukemia relapse. Indeed, PRAME expression has been shown to increase upon progression of CML from chronic-phase to advanced-phase.58 Thus, targeting PRAME in combination with other LAAs would be a rational strategy to eliminate leukemia. The predominance of low-avidity PRAME-specific CD8+ T cells in patients with leukemia could result in expansion of low-avidity T cells after vaccination, with limited efficacy to eradicate leukemia. Nevertheless, it is possible that if sufficient numbers of low-avidity PRAME-specific T cells could be elicited, adequate antitumor activity might still be possible as has been shown for PR1 antigen, where low-avidity T cells were shown to be 2-fold less potent than their high-avidity counterparts at killing CML targets.23 Thus, while our findings encourage us to further explore the potential of PRAME as a leukemia vaccine, these studies reveal possible limitations to the nature of the PRAME immune response to leukemia. Furthermore, achievement of a state of MRD may be required before peptide vaccination to augment T cell immune surveillance. An alternative approach would be the isolation of high-affinity PRAME T-cell receptors from PRAME-specific CTLs generated from healthy donors. T cells can then be transduced with the PRAME-specific T-cell receptors and used in the treatment of patients with PRAME-expressing leukemia.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
We thank Mrs Faith Williams for her technical assistance in the presentation of figures. K.R. acknowledges the support of the National Institute for Health Research (NIHR) Biomedical Research Center.
This work was supported in part by the Kay Kendall Leukemia Fund (grant no. KKL 314).
National Institutes of Health
Authorship
Contribution: K.R. designed and performed experiments, analyzed data, and wrote the manuscript; A.S.M.Y., A.T., and S.M. performed experiments and commented on the manuscript; B.J., R.E., K.K., and Y.L. performed experiments; B.S. advised on statistical analysis and commented on the manuscript; and R.K. and J.B. supervised the laboratory study and commented on the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Katayoun Rezvani, Department of Hematology, Imperial College, Hammersmith Campus, 4th Floor Commonwealth Building, DuCane Road, London W12 0NN, UK; e-mail: k.rezvani@imperial.ac.uk.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal