

Abstract
Because protein function regulates the phenotypic characteristics of cancer, a functional proteomic classification system could provide important information for pathogenesis and prognosis. With the goal of ultimately developing a proteomic-based classification of acute myeloid leukemia (AML), we assayed leukemia-enriched cells from 256 newly diagnosed AML patients, for 51 total and phosphoproteins from apoptosis, cell-cycle, and signal-transduction pathways, using reverse-phase protein arrays. Expression in matched blood and marrow samples were similar for 44 proteins; another 7 had small fold changes (8%-55%), suggesting that functional proteomics of leukemia-enriched cells in the marrow and periphery are similar. Protein expression patterns were independent of clinical characteristics. However, 24 proteins were significantly different between French-American-British subtypes, defining distinct signatures for each. Expression signatures for AML with cytogenetic abnormalities involving −5 or −7 were similar suggesting mechanistic commonalities. Distinct expression patterns for FMS-like tyrosine kinase 3–internal tandem duplication were also identified. Principal component analysis defined 7 protein signature groups, with prognostic information distinct from cytogenetics that correlated with remission attainment, relapse, and overall survival. In conclusion, protein expression profiling patterns in AML correlate with known morphologic features, cytogenetics, and outcome. Confirmation in independent studies may also provide pathophysiologic insights facilitating triage of patients to emerging targeted therapies.
Introduction
The current classification of acute myeloid leukemia (AML) uses the French-American-British (FAB) system based on morphologic features, along with flow cytometric analysis of surface marker expression, cytogenetics, and assessment of recurrent molecular abnormalities. These classification schemes have prognostic relevance, but they generally do not alter therapeutic recommendations.1 Furthermore, current prognostic models, based on clinical and laboratory features, have low predictive power explaining less than half of the outcome. Abnormal activation of signal transduction pathways (STPs), aberrant cell-cycle regulation, and evasion of apoptosis are key events in malignant transformation.2-4 Expression of STP proteins is heterogeneous and prognostic in AML.5-10 Distinct molecular abnormalities and patterns of pathway activation in leukemic cells combine to suggest potential targets for therapeutic intervention. However, unlike chronic myelogenous leukemia, where all cases start with the same chromosomal abnormality, AML is markedly heterogeneous with numerous genetic aberrations. Consequently, knowledge facilitating individualization of targeted therapies under development in AML is sorely needed. Thus, an improved understanding of leukemic cell biology might lead to improved classification schemes that more accurately explain the heterogeneity of response and thereby guide the use of targeted therapies on an individualized basis
Novel array technologies enable the analysis of numerous features at the level of DNA copy number, mutations, methylation, mRNA transcription, regulatory microRNA, and emerging approaches to assess protein expression levels within cells and cytokine and chemokine arrays to assess external forces acting on leukemic cells. Gene expression profiling arrays have demonstrated the ability to provide new classification schema and to define prognostic subgroups in AML.11,12 The contribution of these approaches to patient management and to understanding the pathophysiology of AML remains to be fully elucidated. Transcriptional profiles can provide superior tumor classification, response, and prognostic information. However, RNA transcript levels often do not correlate with protein expression13-15 and differentially expressed genes may not necessarily be involved in the pathogenesis of a disease. Protein expression and posttranslational modifications, either alone or in concert with other profiling approaches, could provide independent or complementary information not captured by transcriptional profiles. Thus, the functional proteome provides an untapped information source, which may contribute prognostic information and provide important pathophysiologic cues regarding response to current or emerging therapies as well as potential new targets for therapy. It is therefore critical to determine whether protein levels and posttranslational modifications can provide novel determinants of cellular phenotype and biologic behavior. Expression levels and activation of single or a few proteins have been studied,5-9,16 but comprehensive analysis of the complexity of intracellular STP is lacking because of lack of a feasible, quantitative, high-throughput proteomic technology applicable to patient samples.
To generate a comprehensive proteomic profile of the level of expression and activation of apoptosis, STP, and cell-cycle regulating proteins in AML, we used reverse-phase protein arrays (RPPA), an approach where protein lysates from multiple samples are printed onto a slide and probed with a single antibody to generate a quantitative output.17-19 Proteins and their corresponding phosphoproteins can be assessed, reflecting the activation state and functionality of a given protein, pathway, or network.20,21 The technique offers high sensitivity, throughput, interslide and intraslide (array) reproducibility, thereby overcoming the limitations of conventional protein assay techniques (Western blotting or enzyme-linked immunosorbent assays [ELISA]).20 The usefulness, high precision, and reliability of RPPA for analysis of leukemic specimens have been demonstrated.18
In this report, we present a set of 320 samples from 256 newly diagnosed, AML patients profiled for 51 total and phosphoprotein epitopes involved in STP, apoptosis, and cell-cycle regulatory pathways. These protein expression signatures provide prognostic information and could potentially guide the selection of targeted therapies.
Methods
Patient population
This study used a total of 539 bone marrow, peripheral blood, or pheresis specimens from 442 patients with AML, including 97 cases with a same-day bone marrow and peripheral blood or pheresis sample These samples were collected at diagnosis (320 samples from 256 patients), in a primary refractory state (59 samples from 47 patients), in relapse (138 samples from 116 patients), in remission (9 samples from 8 patients), and while receiving therapy (n = 7). The same-day bone marrow and peripheral blood or pheresis samples permit comparison of expression between blood and marrow. This report is restricted to the 256 newly diagnosed patients: the associated demographics are described in Table 1. Samples were collected for the Leukemia Sample Bank at the University of Texas M. D. Anderson Cancer Center between January 15, 1998, and March 9, 2006, on institutional review board (IRB)–approved protocol Lab01-473, and consent was obtained in accordance with the Declaration of Helsinki. Samples were analyzed under an IRB-approved laboratory protocol (Lab05-0654).
Demographics and clinical characteristics of the 256 newly diagnosed AML patients in the study set
| Variable category | Number or % |
|---|---|
| No. of cases | 256 |
| % male | 53.8% |
| Race | |
| Asian | 1.9% |
| Black | 8.3% |
| Hispanic | 12.8% |
| White | 76.7% |
| Age, y | |
| Mean | 61.1 |
| Median | 63.8 |
| Minimum | 16.1 |
| Maximum | 87.2 |
| FAB | |
| M0 | 6.5% |
| M1 | 13.8% |
| M2 | 26.8% |
| M4 | 23.4% |
| M4EOS | 3.4% |
| M5 | 2.3% |
| M5A | 5.4% |
| M5b | 3.8% |
| M6 | 2.7% |
| M7 | 1.9% |
| RAEBT | 1.9% |
| Unknown | 8.0% |
| WHO classification | |
| AML with recurrent cytogenetic changes | 29 |
| AML with multilineage dysplasia | 59 |
| AML, therapy related | 36 |
| AML not otherwise categorized | 134 |
| Cytogenetics | |
| Favorable | 8.3% |
| Intermediate | 45.7% |
| Unfavorable | 44.5% |
| ND | 1.5% |
| Zubrod PS | |
| 0-2 | 94.1% |
| 3 or 4 | 5.9% |
| AHD | |
| ≥ 2 mo | 32.1% |
| Response | |
| CR | 56.8% |
| CRP | 3.2% |
| Early death | 0.5% |
| Fail | 8.1% |
| Resistant | 31.1% |
| Inevaluable | 0.5% |
| Relapse | |
| Yes | 54.1% |
| Alive | |
| Yes | 29.4% |
| Variable category | Number or % |
|---|---|
| No. of cases | 256 |
| % male | 53.8% |
| Race | |
| Asian | 1.9% |
| Black | 8.3% |
| Hispanic | 12.8% |
| White | 76.7% |
| Age, y | |
| Mean | 61.1 |
| Median | 63.8 |
| Minimum | 16.1 |
| Maximum | 87.2 |
| FAB | |
| M0 | 6.5% |
| M1 | 13.8% |
| M2 | 26.8% |
| M4 | 23.4% |
| M4EOS | 3.4% |
| M5 | 2.3% |
| M5A | 5.4% |
| M5b | 3.8% |
| M6 | 2.7% |
| M7 | 1.9% |
| RAEBT | 1.9% |
| Unknown | 8.0% |
| WHO classification | |
| AML with recurrent cytogenetic changes | 29 |
| AML with multilineage dysplasia | 59 |
| AML, therapy related | 36 |
| AML not otherwise categorized | 134 |
| Cytogenetics | |
| Favorable | 8.3% |
| Intermediate | 45.7% |
| Unfavorable | 44.5% |
| ND | 1.5% |
| Zubrod PS | |
| 0-2 | 94.1% |
| 3 or 4 | 5.9% |
| AHD | |
| ≥ 2 mo | 32.1% |
| Response | |
| CR | 56.8% |
| CRP | 3.2% |
| Early death | 0.5% |
| Fail | 8.1% |
| Resistant | 31.1% |
| Inevaluable | 0.5% |
| Relapse | |
| Yes | 54.1% |
| Alive | |
| Yes | 29.4% |
RAEBT indicates refractory anemia with excess of blasts in transformation; AML, acute myeloid leukemia; AHD ≥ 2, antecedent hematologic disorder of greater than or equal to 2 months; CR, complete remission; CRp, complete remission criteria met except that platelets fail to reach 100 000 μL; Early death, death within the first 2 weeks after the start of induction therapy; and Fail, death occurring more than 2 weeks after the start of therapy.
Among the 256 newly diagnosed, previously untreated (new) patients, 120 were treated with anthracycline and high-dose ara-C (HDAC)–based regimens, 49 on HDAC plus nonanthracycline chemotherapy regimens (eg, fludarabine, HDAC, G-CSF [FLAG], fludarabine + HDAC [FA], clofarbine), 21 with other non–HDAC-based chemotherapy, 10 with targeted agents, 10 with gemtuzumab ozogamicin (Mylotarg)-based regimens, 6 with demethylating or histone-deacetylating agents, 1 with low-dose ara-c, one with stem-cell transplant as initial therapy, and 40 not treated at M. D. Anderson Cancer Center.
Specimen collection and processing
Immediately after collection, samples were placed on ice and processed within 2 hours by Ficoll-Hypaque gradient separation to yield a mononuclear cell fraction. The isolated mononuclear cell fraction containing blasts was further enriched by depletion of CD3+CD19+ B and T lymphocytes using magnetic antibody-conjugated sorting (Miltenyi Biotec, Auburn, CA) as previously described.7 The purity of the leukemic samples was generally greater than 80% blasts after enrichment. After isolation and washing, boiling hot protein lysis buffer (2×, 0.5 M Tris-HCl, pH 6.8, 2% sodium dodecyl sulfate, 10% glycerol, and 4% β-mercaptoethanol) was added to make a whole-cell lysate, which is then aliquoted into single-use vials before cryopreservation.5,6 We have previously demonstrated that protein made from cryopreserved AML cells yield identical results on RPPA compared to protein made from fresh cells on the day of collection, demonstrating that protein levels are not adversely affected by cryopreservation of cells or protein lysates.18
Controls, reference standard, and cell lines
To correct for staining, background, and loading variation across (array) slides, a positive control consisting of a mixture of 11 cell lines hereafter called “pooled control” lysate and the lysate buffer as negative control were used. For expression controls, 18 cell lines, including baseline and growth factor or cytokine-stimulated samples, were used and shown on the schematic by the letter “C” (Figure 1A). Eighteen peripheral blood samples from healthy volunteers (IRB-approved collection) served as normal controls, shown by the letter “N” in Figure 1. To permit absolute quantification of signal strength, we included 138 purified peptides from sequences used to generate antibodies in the study as well as additional controls, shown by the purple band encircling the patient samples.
![Figure 1. Array assembly, printing method, topographic normalization, and example slide. (A) Each letter represents a group of specimens. Replicates of the same patient sample were printed in area “A” and in a reversed orientation in area “B.” As controls, 18 cell lines (MDA-MD-231, MDA-MD-231 stimulated with IGF, MDA-MD-468, MDA-MD-468 stimulated with EGF, Hela HL-60, Jurkat [× 2 from separate sources], Jurkat incubated with anti-FAS antibody, K562, Kasumi-1, MV4-11, NB4, OCIAML3, Raji, THP-1, U937, Y79) were printed in area “C” and 18 normal peripheral blood samples in area “N.” Purified peptides (n = 138) were printed encircling the patient samples, as shown by the purple band. (B) Schematic showing the dilution series of patient samples. At the end of each row of patient samples, a positive control made of a mixture of 11 cell lines (Hela, HL60, Jurkat, Kasumi1, K562, KG1, MV4-11, OCI-AML3 NB4, U937, Y79) (pooled control, shown as a red “+”) or negative control (protein lysis buffer, shown as a “−”) is printed, creating a grid across the whole slide for negative and positive controls to permit topographic normalization. (C) Representative slide of phospho-AKT (Thr308). The overlays in purple, red, and green covers the areas where the purified peptides, cell lines, and normal peripheral blood samples and positive and negative control are located as depicted in Figure 1A. The insert demonstrates phosphorylated and unphosphorylated AKT peptide specifically detected by the antiphospho-AKT (Thr308) antibody. (D,E) The 3-dimensional (D) topographic map of the negative control generates a 3-dimensional topographic map that can be used to correct for background, whereas the 3-dimensional grid of the positive controls sets the scale for quantification. A topographic map from a slide (probed for BAK) with low background that is even across the slide (D) and another (E) from the slide (probed for MCL1) with the most background variation across the slide are shown.](/view-large/figure/6954078/zh80240828470001.jpeg)
Array assembly, printing method, topographic normalization, and example slide. (A) Each letter represents a group of specimens. Replicates of the same patient sample were printed in area “A” and in a reversed orientation in area “B.” As controls, 18 cell lines (MDA-MD-231, MDA-MD-231 stimulated with IGF, MDA-MD-468, MDA-MD-468 stimulated with EGF, Hela HL-60, Jurkat [× 2 from separate sources], Jurkat incubated with anti-FAS antibody, K562, Kasumi-1, MV4-11, NB4, OCIAML3, Raji, THP-1, U937, Y79) were printed in area “C” and 18 normal peripheral blood samples in area “N.” Purified peptides (n = 138) were printed encircling the patient samples, as shown by the purple band. (B) Schematic showing the dilution series of patient samples. At the end of each row of patient samples, a positive control made of a mixture of 11 cell lines (Hela, HL60, Jurkat, Kasumi1, K562, KG1, MV4-11, OCI-AML3 NB4, U937, Y79) (pooled control, shown as a red “+”) or negative control (protein lysis buffer, shown as a “−”) is printed, creating a grid across the whole slide for negative and positive controls to permit topographic normalization. (C) Representative slide of phospho-AKT (Thr308). The overlays in purple, red, and green covers the areas where the purified peptides, cell lines, and normal peripheral blood samples and positive and negative control are located as depicted in Figure 1A. The insert demonstrates phosphorylated and unphosphorylated AKT peptide specifically detected by the antiphospho-AKT (Thr308) antibody. (D,E) The 3-dimensional (D) topographic map of the negative control generates a 3-dimensional topographic map that can be used to correct for background, whereas the 3-dimensional grid of the positive controls sets the scale for quantification. A topographic map from a slide (probed for BAK) with low background that is even across the slide (D) and another (E) from the slide (probed for MCL1) with the most background variation across the slide are shown.
Array assembly, printing method, topographic normalization, and example slide. (A) Each letter represents a group of specimens. Replicates of the same patient sample were printed in area “A” and in a reversed orientation in area “B.” As controls, 18 cell lines (MDA-MD-231, MDA-MD-231 stimulated with IGF, MDA-MD-468, MDA-MD-468 stimulated with EGF, Hela HL-60, Jurkat [× 2 from separate sources], Jurkat incubated with anti-FAS antibody, K562, Kasumi-1, MV4-11, NB4, OCIAML3, Raji, THP-1, U937, Y79) were printed in area “C” and 18 normal peripheral blood samples in area “N.” Purified peptides (n = 138) were printed encircling the patient samples, as shown by the purple band. (B) Schematic showing the dilution series of patient samples. At the end of each row of patient samples, a positive control made of a mixture of 11 cell lines (Hela, HL60, Jurkat, Kasumi1, K562, KG1, MV4-11, OCI-AML3 NB4, U937, Y79) (pooled control, shown as a red “+”) or negative control (protein lysis buffer, shown as a “−”) is printed, creating a grid across the whole slide for negative and positive controls to permit topographic normalization. (C) Representative slide of phospho-AKT (Thr308). The overlays in purple, red, and green covers the areas where the purified peptides, cell lines, and normal peripheral blood samples and positive and negative control are located as depicted in Figure 1A. The insert demonstrates phosphorylated and unphosphorylated AKT peptide specifically detected by the antiphospho-AKT (Thr308) antibody. (D,E) The 3-dimensional (D) topographic map of the negative control generates a 3-dimensional topographic map that can be used to correct for background, whereas the 3-dimensional grid of the positive controls sets the scale for quantification. A topographic map from a slide (probed for BAK) with low background that is even across the slide (D) and another (E) from the slide (probed for MCL1) with the most background variation across the slide are shown.
Array assembly and printing method
For quantification purposes, 5 serial dilutions (1:3 dilution steps) of each protein lysate were arrayed in 384-well plates (Genetix, Boston, MA). Samples were printed onto nitrocellulose-coated glass slides (FAST Slides; Whatman Schleicher and Schuell, Keene, NH) using an Aushon Biosytems 2470 Arrayer (Aushon BioSystems, Burlington, MA) with 175-μm pins and a single touch. The samples were printed in replicate, one centrally located and the other split on either side and arranged in a reversed orientation (Figure 1A). Based on the sample concentration of 1 × 104 cells per microliter and a printing volume of 2 nL/touch, we estimate that the spots ranged from 85 cell equivalents of protein in undiluted, with approximately 1 cell protein equivalent in the most diluted (1:81) spot (Figure 1B). To permit topographic normalization, a sample of pooled or negative control sample was printed at the end of each row of patient sample, creating a grid across the whole slide, which appears as alternating spots in the sample array (Figure 1C). Each slide contained 7968 dots. A representative slide is shown in Figure 1C.
Antibody detection and array staining
A detailed description of the array methodology, including antibody staining and detection, has been published.18 Briefly, after printing, slides were incubated for 15 minutes in biotin blocking solution to block endogenous peroxidase, avidin, and biotin before incubating slides in protein block at 4°C overnight. Primary antibodies in concentrations from 1:250 to 1:2000 were added for 1 to 2 hours with frequent rotation (dilutions and manufacturers of antibodies, Table S1, available on the Blood website; see the Supplemental Materials link at the top of the online article). A biotinylated secondary antibody (antimouse or antirabbit), diluted 1:10 000 to 1:20 000, used as starting point for signal amplification, was added for 1 hour. Subsequently, array slides were incubated using the Dako Denmark (Glostrup, Denmark) signal amplification system using catalyzed reporter deposition of substrate to amplify the signal detected by the primary antibody.22 Slides were incubated in streptavidin-biotin-peroxidase and biotinyl-tyramide/hydrogen peroxide reagents each for 15 minutes with frequent washing in between. Finally, 3,3′-diaminobenzidine tetrachloride was cleaved by tyramide-bound horseradish peroxidase, giving a stable brown precipitate with excellent signal-to-noise ratio. This technique is sensitive and reproducible to the femtomolar range as reported.18,23
Fifty-one proteins were assayed, including 30 total and 21 phosphoproteins (Table S1), using antibodies validated as described previously.18 We have used a naming system where the protein name always comes first, followed by description of any modification (eg, “p” for phosphorylation), followed by the amino acid site of the modification when more than one phosphorylation site was analyzed (eg, AKT.p308 vs AKT.p473). This differs from the convention of using a lowercase “p” before the protein name. This system facilitates alphabetical sorting and provides additional detail regarding modification site. Many of these same samples (n = 125) were used in our prior study looking at STP expression in AML by Western blot.7 There was significant correlation between the RPPA and the Western blot results for ERK(P = .03), ERK.p (P = .003), AKT.p473 (P = .02), and PKCα (P = .04) using the same antibodies.
Data analysis, normalization, and statistics
Hybridized slides were scanned on a desktop scanner (Hewlett Packard, Palo Alto, CA) at an optical resolution of 1200 dpi in 16-bit grayscale and saved as TIFF files. Protein expression intensity of each spot was measured with an automated software program MicroVigene (VigeneTech, North Billerica, MA). The dilution series of the samples provide a dilution-concentration-expression curve providing relative expression intensities. These numbers were used for data processing and calculation after standardization and topographic normalization. All analyses were performed using the R Statistical Programming Environment, version 2.4.0.
Topographic normalization
Positive and negative controls were positioned across the slide at the end of each sample as described in “Array assembly and printing method.” This enables normalization to adjust for variations in background staining. Negative controls (lysis buffer) set the baseline noise for background correction and positive controls (cell lines, peptides) provide expression intensities to set the scale for protein quantification in the samples. Background variation was variable, as shown in the topographic maps ranging from negligible to the most extreme (Figure 1D,E). After subtracting local background estimates from the MicroVigene quantifications, we subtracted the median background-corrected values of the negative controls from all spots. This ensured that the negative controls were centered around zero and resulted in data where replicate spots yielded more reproducible values and where the slope of the linear part of the concentration curves more closely matched the known dilution step. Next, the median intensity, M, of the positive controls was computed, and each dilution series was normalized by dividing by M times the intensity of the nearest positive control. Other processing methods were evaluated, but this procedure yielded superior results, resulting in the highest correlation (R2 > 0.8) between the replicates on a slide. Correlation between sample 1 and sample 2 on the slide was very high with R more than 0.7 for 59%, more than 0.5 in 92% of the proteins studied after topographic background correction. Neeley et al (S. Neeley, K. A. Baggerly, S.M.K., manuscript submitted) provide a more detailed description of the algorithm. The average multipliers for correction for loading and regional staining usually ranged between 0.624 and 1.376.
Summarization of dilution series
After preprocessing, dilution series were summarized by fitting a joint 4-parameter logistic model as described by Tabus et al.24 We used our own implementation of this algorithm in an R package, SuperCurve 0.931 (http://bioinformatics.mdanderson.org/Software/OOMPA).
Various clustering methods were used. Validity of clusters was assessed using perturbation bootstraps25 and the gap statistic.26 Associations between protein expression levels and categorical clinical variables were assessed in R using standard t tests, linear regression, or mixed-effects linear models. Conservative Bonferroni corrections were performed to account for multiple statistical parameters (numbers of samples and proteins/antibodies) when calculating statistical significance.
The dataset is available at http://bioinformatics.mdanderson.org/supplements.html (under “RPPA Data in AML”) or as an Excel file from skornblau@mdanderson.org.
Results
Blood and marrow specimens can be used interchangeably
This array contained same-day blood/pheresis and marrow samples from 97 patients, permitting a direct comparison of protein levels in blast cells from blood and marrow. If expression was significantly different depending on the source, then separate analysis would be required for blood and marrow. In contrast, if they were similar, then all the samples could be combined into a single analysis. Similar to our previous observations in Western blot27 and RPPA,18 linear mixed effects models revealed no statistically significant differences in expression intensities between blood and marrow for most (44 of 51) proteins. However, 7 proteins had statistically significant differences (Table 2): 3 were higher in blood and 4 higher in marrow. Although these differences were statistically significant after Bonferroni correction, 4 had absolute fold differences less than 20%, making it doubtful that these differences have biologic relevance. The others, SRCp527, S6RP.p228, and S6RP.p240-244, had fold differences ranging from 28% to 55%, which might reflect proliferation potential (“Discussion”). Consequently, blood and marrow-derived samples were combined for the subsequent analysis. Replicate measurements from the same patient were averaged, with the 7 proteins where blood and marrow levels differed being adjusted to remove this small effect.
Proteins with different expression between blood and marrow
| Protein | P | Fold |
|---|---|---|
| Blood higher | ||
| BAK | 2.5 × 10−4 | 1.082 |
| BAD | 1.3 × 10−4 | 1.113 |
| SRCp527 | 5.5 × 10−8 | 1.278 |
| Marrow higher | ||
| Survivin | 3.8 × 10−7 | 1.113 |
| MTOR.p | 8.5 × 10−4 | 1.123 |
| S6RP.p235 | 2.8 × 10−7 | 1.400 |
| S6RP.p240–244 | 1.0 × 10−16 | 1.553 |
| Protein | P | Fold |
|---|---|---|
| Blood higher | ||
| BAK | 2.5 × 10−4 | 1.082 |
| BAD | 1.3 × 10−4 | 1.113 |
| SRCp527 | 5.5 × 10−8 | 1.278 |
| Marrow higher | ||
| Survivin | 3.8 × 10−7 | 1.113 |
| MTOR.p | 8.5 × 10−4 | 1.123 |
| S6RP.p235 | 2.8 × 10−7 | 1.400 |
| S6RP.p240–244 | 1.0 × 10−16 | 1.553 |
Correlation of protein profiles with clinical characteristics
No correlation between individual protein expression levels and traditional clinical characteristics (eg, age, sex, race, infection status, performance status, antecedent hematologic disorder, history of prior malignancy, radiation or chemotherapy, white blood cell count, platelet count, hemoglobin, creatinine, bilirubin, albumin, percent blood and marrow blasts, percent cells expressing CD 7, 10, 13, 19, 20, 33, 34, cytogenetics, and treatment regimen) was observed (data not presented).
Correlation of protein profiles with FAB subtypes
Because the FAB classification distinguishes leukemias based on differentiation pathways and the degree of maturation, we speculated that there might be unique protein expression signatures for different FAB types. We used linear regression, with individual proteins, to perform a one-way analysis of variance (ANOVA) with Bonferroni correction and observed that 24 proteins showed significant heterogeneity in protein expression levels between FAB classes. Because the ANOVA does not describe which FAB classes have significantly different expression levels for a protein, we used Tukey's test for honestly significant differences to determine which mean differences between classes were statistically significant. Next, we clustered the significant proteins based on their patterns of protein expression across the newly diagnosed AML patients and produced a heatmap of their (standardized) average expression by FAB category (Figure 2). We found 3 clusters of proteins that tended to track similarly within an FAB class. The first cluster was enriched for total and phosphorylated signal transduction proteins (PKCA, PKCA.p, ERK2, AKT.p308, P38.p P70S6K, P70S6K.p, and Src.p527); proteins in this cluster were primarily characterized by lower expression in M0, M1, M2, with higher levels in the other FAB types. The differences between early myeloid (M0, M1, M2) were statistically significant different in comparison to FAB subtypes with a monocytic component (M4, M5). The second cluster contains PTEN and PTEN.p; it is characterized by significantly lower expression in M6 and M7. The third cluster is enriched for apoptosis (BAD, BAK, BCL2, BCLXL, BAD.p136, SMAC), cell cycle or differentiation regulating proteins (Myc, CCND1, SSBP2), and activated STAT proteins. In general, these proteins have significantly higher expression in the myeloid subtypes (M0, M1, and M2) compared with the monocytic (M4, M5), erythroid (M6), or megakaryocytic (M7) subtypes. Within this group, β-catenin (BCAT), TP27, and BCL-XL exhibit a slightly different pattern, with significantly higher expression in M6 or M7.

Hierarchical clustering of antibodies (AB) differentially expressed among FAB groups. The heatmap graphically represents the 24 AB whose expression levels were significantly different between the FAB subgroups. The number of patients in each group is shown along the bottom of the figure. Protein levels were normalized such that the expression level of M1 was set to 0 and the expression of all other proteins is shown as fold change from this baseline. Each protein was standardized so that each protein has mean 0 and SD 1 across the categories. Expression is scaled so that green represents low expression and red represents high expression.
Hierarchical clustering of antibodies (AB) differentially expressed among FAB groups. The heatmap graphically represents the 24 AB whose expression levels were significantly different between the FAB subgroups. The number of patients in each group is shown along the bottom of the figure. Protein levels were normalized such that the expression level of M1 was set to 0 and the expression of all other proteins is shown as fold change from this baseline. Each protein was standardized so that each protein has mean 0 and SD 1 across the categories. Expression is scaled so that green represents low expression and red represents high expression.
Expression patterns and cytogenetics
Recurrent cytogenetic abnormalities currently provide the strongest available predictors of therapy response and outcome in AML.28 To define the relationship between protein expression and cytogenetic groups, we computed the average protein profile for each cytogenetic profile, then clustered these profiles using complete linkage and Pearson correlation (Figure 3). Based on the gap statistic,26 there are 5 clusters in the data: (1) a cluster driven by changes involving chromosomes 5 or 7, (2) Ph1, (3) t(8;21), 11q23, and IM, (4) the complex karyotype of simultaneous −5, −7, and 11q23, and (5) a miscellaneous cluster including diploid, t(6;9), inv(16), +8, and +21. AMLs with cytogenetic aberrations are associated with distinct protein patterns that differ from the mean. In contrast, the diploid and miscellaneous AMLs show an “average” rather than a specific expression pattern, probably reflecting heterogeneity in these cases.

Clustering by cytogenetics average protein expression intensity of patients within cytogenetic risk groups based on unsupervised hierarchical clustering. Expression is scaled so that green represents low expression and red represents high expression.
Clustering by cytogenetics average protein expression intensity of patients within cytogenetic risk groups based on unsupervised hierarchical clustering. Expression is scaled so that green represents low expression and red represents high expression.
As with the FAB subtypes, we performed gene-by-gene ANOVAs to determine which individual proteins were differentially expressed in patients with different cytogenetic abnormalities. After Bonferroni correction, only 2 proteins were found to be significantly different: Tp53 (P = 1.32 × 10−13) and BAX (4.73 × 10−7) across all groups. Tp53 was significantly higher whenever there was an abnormality of chromosome 5 or chromosome 7. BAX, by contrast, had 3 distinct levels of expression: low when there were abnormalities of 5 or 7, intermediate in 11q23 (even in the presence of losses of 5 and 7), inv(16), diploid, or miscellaneous samples, and high in all other categories. In addition, cases with a FMS-like tyrosine kinase 3–internal tandem duplication (FLT3-ITD) were observed to have significantly higher expression of pMTOR (P < .001), pStat5 (P < .001) AKT, Badp136, BAX, MEK, MTOR, pPTEN, pStat1(P < .01) and MYC, PKCα, pPKCα, SRCp527 (P < .05). Association between FLT3-ITD and Stat5 phosphorylation29,30 and PKCα mRNA expression31 has been previously described.
Principal component analysis and protein signatures groups
We hypothesized that proteins would form “constellations” in which the components provided predictive information. The most commonly used similarity measures (Euclidean distance or Pearson correlation) treat negatively correlated variables as highly distinct. To deal with this issue, we clustered the proteins using a distance d = (1 − |ρ|) based on the absolute value of the Pearson correlation, ρ. This clustering suggested the existence of 10 protein constellations (CNSTN; Figures 4, S2). In many constellations, the proteins have related functions supporting the idea that the approach has functional validity. For example, CNSTN-9 contain phosphorylated signaling pathway members TP38.p, MEK.p, ERK.p, and inactivating phosphorylation of 2 of their targets, GSK3.p and BAD.p112; CNSTN-6 contains the cell survival signaling proteins AKT.p308 and AKT.p473 inversely correlated with apoptosis regulating proteins BLC2, Bad, and SMAC; CNSTN-1 has apoptosis-related proteins survivin, XIAP, and Bak in a positive correlation with cyclinD1 and negative correlation with phosphorylation of S6, which would promote transcription and proliferation; and CNSTN-4 has phosphorylated forms of STAT1, 5, and 6. Other constellations contain proteins active in separate pathways that might have overlapping functions; eg, CNSTN-8 has both p53 and MCL1, which might be expected to affect apoptosis via separate mechanisms.

Protein expression against SGs. Proteins were clustered into 10 constellations shown by the 10 colored boxes along the left axis and the yellow lines surrounding each constellation. Based on the score for each constellation for each patient, an overall score was generated, and this divided patients into 7 protein SGs (bottom of heatmap). The average score for each protein constellation within each SG is shown. An additional heatmap showing the expression of each protein in each individual patient is available online as Figure S2. The fold change between −2 SDs and 2 SDs for each individual protein, corresponding to the −2 (green) to +2 (red) color gradient bar shown in the upper left corner, is listed to the right of the protein name.
Protein expression against SGs. Proteins were clustered into 10 constellations shown by the 10 colored boxes along the left axis and the yellow lines surrounding each constellation. Based on the score for each constellation for each patient, an overall score was generated, and this divided patients into 7 protein SGs (bottom of heatmap). The average score for each protein constellation within each SG is shown. An additional heatmap showing the expression of each protein in each individual patient is available online as Figure S2. The fold change between −2 SDs and 2 SDs for each individual protein, corresponding to the −2 (green) to +2 (red) color gradient bar shown in the upper left corner, is listed to the right of the protein name.
To integrate and summarize the information from negatively and positively correlated proteins within a constellation, we performed a principal components analysis on each constellation retaining the first principal component. For each constellation and patient, this method computes a score, which is a weighted sum of protein levels, which describes the extent to which the constellation is present in the protein expression pattern of that patient. For example, in the first principal component from CNSTN 7, the proteins SRC and SRC.p527 get negative weights (−0.83 and −0.47, respectively), whereas S6 and SSBP3 get positive weights (0.10 and 0.27, respectively). We then clustered the patients, using Pearson correlation and complete linkage, based on these scores. Based on the gap statistic, the scores separate the patients into 7 protein signature groups (SGs), although there is some indication that the high level split into 2 groups (group A = SG1, SG2, and SG6; group B = SG3, SG4, SG5, and SG7) gives the most reliable separation (Figure 4). The heatmap showing the average normalized expression for each protein, within each SG, shows that in some SGs all proteins within a particular constellation are positively correlated (eg, CNSTN-5, the GSK3, BCL-XL, TP27, BCAT, DJI group), whereas in others some proteins are positively correlated with some members and negatively correlated with others (eg, CNSTN-6 with P70S6K.p, P70S6, AKT.p308, AKT.p473, which correlate with each other, but are negatively correlated with other constellation members BAD, SMAC, SSBP2, BCL2). This arrangement allows for better visualization of relative over and underexpression proteins within each of the 10 protein constellations within each of the 7 SG.
The distribution of patients with favorable, intermediate, or unfavorable cytogenetics among the 7 SG was signi-ficantly uneven (χ2 test, = 40.63 on 12 degrees of freedom, P = 5.66 × 10−5; Table 3). Patients with favorable cytogenetics were significantly overrepresented in SG2 and underrepresented in SG7. Patients with intermediate cytogenetics were overrepresented in SG3 and SG6 and underrepresented in SG5. Patients with unfavorable cytogenetics were overrepresented in SG5 and SG7 and underrepresented in SG3.
Distribution of cytogenetic risk groups
| SG group | Observed (expected) count | ||
|---|---|---|---|
| Favorable | Intermediate | Unfavorable | |
| 1 | 1 (2.83) | 19 (15.84) | 14 (15.33) |
| 2 | 12 (4.17)* | 19 (23.30) | 19 (22.54) |
| 3 | 4 (3.42) | 25 (19.10)* | 12 (18.48)* |
| 4 | 2 (3.25) | 19 (18.17) | 18 (17.58) |
| 5 | 1 (2.67) | 8 (14.91)* | 23 (14.42)* |
| 6 | 2 (2.92) | 21 (16.31)* | 12 (15.78) |
| 7 | 0 (2.75)* | 12 (15.38) | 21 (14.88)* |
| SG group | Observed (expected) count | ||
|---|---|---|---|
| Favorable | Intermediate | Unfavorable | |
| 1 | 1 (2.83) | 19 (15.84) | 14 (15.33) |
| 2 | 12 (4.17)* | 19 (23.30) | 19 (22.54) |
| 3 | 4 (3.42) | 25 (19.10)* | 12 (18.48)* |
| 4 | 2 (3.25) | 19 (18.17) | 18 (17.58) |
| 5 | 1 (2.67) | 8 (14.91)* | 23 (14.42)* |
| 6 | 2 (2.92) | 21 (16.31)* | 12 (15.78) |
| 7 | 0 (2.75)* | 12 (15.38) | 21 (14.88)* |
The 3 major cytogenetic risk groups are unevenly distributed across the seven protein signature groups. Each cell contains the observed (and expected under the null hypothesis) number of patients in each group.
Observations that differed most from the expected values (and thus contributed significantly to the χ2 test).
Effect of SG on remission, relapse, and survival
Although the complete response (CR) rates within the 7 SG ranged from 42% to 73%, these differences were not statistically significant by χ2 test (8.04 on 6 degrees of freedom, P = .24). Rather than combine groups arbitrarily, we analyzed the data using a hierarchical Bayesian model. The response within SGi was assumed to be binomial with rate θi. The response rates were assumed to arise from a common beta distribution θi ∼ Beta(α, β). We used an uninformative prior on the natural transformed scale:
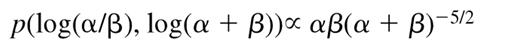
For details on the model, see section 5.3 of Gelman et al.32 We found that SG7 (which is enriched for unfavorable cytogenetics and depleted of favorable cytogenetics, with an overall CR rate of 42%) has a posterior probability of at least 80% of having a lower CR rate than SG2, SG3 SG4, or SG6, and at least a 69% posterior probability of having a lower CR rate than SG1 or SG5. We also found that SG2 (which is enriched for favorable cytogenetics, with an overall CR rate of 72%) has a posterior probability of at least 70% of having a higher CR rate than every other group except for SG6. The relapse rates, which ranged from 37% in SG3 to 75% in SG6 were not statistically significantly different by χ2 test (7.79 on 6 degrees of freedom, P = .25).
Next, we looked at associations between the SG and overall survival. Using a Cox proportional hazards model, we found a significant difference in overall survival (log-rank test; score = 16.6 on 6 degrees of freedom; P = .011; Figure 5A). We found that patients in SGs SG2, SG3, and SG4 had the best survival (hazard ratios, 1 ≤ HR ≤ 1.28), whereas patients in SG5 (HR = 2.29), SG7 (HR = 2.14), and SG6 (HR = 2.13) had the worst survival.

Overall survival in SGs in all patients and in diploid FLT3-ITD cases. Kaplan-Meier estimates of proportion of surviving patients over time in weeks for (A) all cases and (B) cases with diploid cytogenetics and a FLT3-ITD.
Overall survival in SGs in all patients and in diploid FLT3-ITD cases. Kaplan-Meier estimates of proportion of surviving patients over time in weeks for (A) all cases and (B) cases with diploid cytogenetics and a FLT3-ITD.
Having observed that SGs were associated with cytogenetics, we also evaluated a Cox proportional hazards model that used both cytogenetics and SGs as predictors of survival. In the joint model, cytogenetics remained highly significant (P = 4.18 × 10−7), whereas the sample group showed weaker evidence of significance (P = .13). When stratified by cytogenetics, the response rate varied by SG for patients with intermediate (range, 40%-67%) and unfavorable cytogenetics (range, 30%-75%; Table 4), but not for those with favorable cytogenetics where all patients achieved remission. For all 3 cytogenetic groups, the relapse rates differed within the different SG (Table 4). As might be expected, these SG-based differences in remission and relapse rates combined to result in different survival outcomes when the effect of SG on survival within each cytogenetic group was determined. Within cytogenetics categories, SG was not significant However, among FLT3-ITD–positive diploid cases, the protein expression signature was still prognostic (P < .001) with patients in SG2 having the best survival, SG3 having intermediate survival, and the other groups having extremely poor survival (Figure 5B).
Complete remission and relapse rate by signature group stratified by cytogenetic risk groups
| SG group | Favorable, % CR/relapse | Intermediate, % CR/relapse | Unfavorable, % CR/relapse |
|---|---|---|---|
| 1 | 100/0 | 67/30 | 30/100 |
| 2 | 100/33 | 65/55 | 54/43 |
| 3 | 100/50 | 67/31 | 33/67 |
| 4 | 100/0 | 67/70 | 47/43 |
| 5 | 100/100 | 40/50 | 53/67 |
| 6 | 100/100 | 60/75 | 75/67 |
| 7 | — | 58/29 | 32/83 |
| SG group | Favorable, % CR/relapse | Intermediate, % CR/relapse | Unfavorable, % CR/relapse |
|---|---|---|---|
| 1 | 100/0 | 67/30 | 30/100 |
| 2 | 100/33 | 65/55 | 54/43 |
| 3 | 100/50 | 67/31 | 33/67 |
| 4 | 100/0 | 67/70 | 47/43 |
| 5 | 100/100 | 40/50 | 53/67 |
| 6 | 100/100 | 60/75 | 75/67 |
| 7 | — | 58/29 | 32/83 |
The 3 major cytogenetic risk groups are unevenly distributed across the seven signature groups. Values are the percentage of patients that have achieved complete remission/the percentage that relapsed.
SG indicates signature group; and —, not applicable.
Discussion
This study has demonstrated that proteomic profiling of AML provides significant details about AML that complement and expand on known classification and prognostic information. We used RPPA because of its high sensitivity, quantitation, and high- throughput capabilities. The sensitivity of RPPA is evident by the ability to frequently see signal from the fifth dilution, corresponding to the protein equivalent of a single cell. The high-throughput capability is demonstrated by the large number of samples analyzed on a single slide (N = 539) and the fact that the 51 slides were stained, scanned, and analyzed in a 10-day period. Several observations emerge from this dataset.
Protein levels in AML cells do not appear to exhibit biologically relevant differences between blood and marrow-derived specimens for most proteins. The presence of 97 same-day blood and marrow specimens permitted direct comparison of expression in these 2 compartments. The majority of proteins examined (44 of 51) had statistically indistinguishable levels of expression. The fold differences for BAD, BAK, MTOR.p, and survivin, although statistically different, are sufficiently modest (range, 1.08-1.20) that they probably don't have biologic impact. There may be biologic relevance to the observations that levels of SRC.p527, an inactivating phosphorylation, are 28% higher in blood, and levels of activated S6RP, phosphorylated on amino acids 235 or 240 to 244, was 40% to 55% higher in marrow. Together, these suggest that circulating cells are in a lower proliferative and transcriptional state compared with marrow bound leukemic blasts. This finding supports our previous observations in 8 proteins using Western blotting27 and by others with gene-expression profile.11 The similarity between marrow and circulating cells indicates that either can be used for proteomic analysis of AML provided that similar methodology is used to generate a leukemia-enriched fraction. For patients with circulating leukemic blasts, this could obviate the need to undergo bone marrow aspiration with its associated procedure costs and discomfort.
Proteomic profiling identified differences between subtypes of AML in the FAB classification schema, which classifies AML, based on lineage differentiation and maturation. A subset of 24 proteins showed differences in expression across the FAB classifications, and the patterns of expression easily distinguish purely myeloid subtypes M0, M1, and M2 from M4 and M5 subtypes with monocytic components, and all of these were distinct from erythroleukemia and megakaryocytic leukemia. These data might be used to suggest treatment targets and guide selection of targeted therapies in different FAB subtypes. For example, expression of phospho-Stat 1 and 5 and several apoptosis-related proteins was higher in M0 to M2 subtypes relative to M4 and M5. In contrast, expression levels of phosphorylated signal transduction proteins AKT, TP38, PKCα, and SRC.p527 were higher in the monocytic subtypes. These patterns suggest that chemoresistance within FAB M0 to M2 leukemias may be more dependent on resistance to apoptosis, whereas growth and resistance to therapy in M4/M5 subtypes might be more dependent on the proliferative and antiapoptotic signals derived from activation of STP.
Similarly, different patterns of protein expression distinguished the major recurrent cytogenetic abnormalities observed in AML. Historically, patients with abnormalities on chromosome 5 and 7 have similar poor response rates and survival with current therapy. Hierarchical clustering of proteomic data demonstrated that −5, 5q−, and −7 or the combinations of −5 and −7 or −5, −7, and +8, had similar protein expression patterns. A similar clustering of −5 and −7 were observed with gene-expression profile.12 Thus, it appears that the different cytogenetic changes have similar effects on protein expression and activation within AML blasts. Dissecting the protein patterns and subjecting them to pathway analysis may yield direct insight into the transforming mechanism(s) underlying these cytogenetic abnormalities. Of note is that p53 expression was highest in these patients, a group shown to have the highest rates of p53 mutations in AML,33 suggesting that use of agents targeting p53 activity or expression, such as MDM2 agonists,34 might be active in these patients. Other examples are a generally higher survivin and GSK3 expression in AML with −5, −7 abnormalities. Survivin is a marker of poor prognosis in advanced stages of AML.35 Survivin inhibitors might be best tested in this group (chromosome 5 and 7 abnormalities) first. Despite having favorable cytogenetics, many patients with inv(16) or t(8:21) relapse and die. Differences in protein expression patterns between long-term survivors and those that ultimately relapse may reveal the underlying differences that result in this dichotomous outcome.
The third major observation is that the overall pattern of protein expression can be used to classify patients into 7 signatures groups based on the summation of expression for each of 10 separate constellations of related protein expression. These constellations consist of proteins that correlate most closely with each other and suggest certain phenotypes for the cells. For example, CNSTN 9 has phospho-ERK, MEK, TP38, and inactivating phosphorylation of BAD and GSK3, a combination of features that promote growth and survival. Signature groups 4, 5, and 7 contain higher levels of CNSTN-8 proteins p53, MCL1, and neuropilin-1. Mutations of p53 are rare in AML, occurring in approximately 5% to 10%36 but correlate with worse outcome.37 Approximately 10% of the samples analyzed had very strong p53, and sequencing revealed a high rate of p53 mutation in high expressers and confirmed the worse prognosis associated with high p53 expression38 (S. Neeley, K. A. Baggerly, S.M.K., K.R.C., manuscript submitted). The increased expression of MCL1 is associated with therapy resistance in AML.39 High expression of Neuropilin-1, which functions in vascular endothelial growth factor and semaphorin receptor signaling as a mediator of angiogenesis, has also been demonstrated to confer an adverse prognosis in AML,8 possibly establishing a vascular endothelial growth factor–dependent autocrine loop that promotes survival.40 The RPPA method thus identifies the combination of mutant p53 and high levels of MCL1 and NRP1 as a novel adverse prognostic combination associated with the lower remission rates, highest relapse, and worse survival. This provides a testable hypothesis that combined therapy directed at restoring p53 function (such as with an MDM2 antagonist),34 inhibiting the antiapoptotic effect of MCL1 and interfering with vascular endothelial growth factor signaling through NRP141 would be beneficial in this subgroup.
In most of the SGs, the protein expression patterns of STP conform to canonical expectations based on the established pathways. For example, in all SGs, the levels of pAKT and pS6RP were well correlated with their respective known substrates GSK342 or P70S6K. Consistent with our prior Western blot findings, we again observed that: there was evidence of crosstalk within STP as levels of PKCα.p, AKT.p308, MEK.p, ERK.p, and TP38.p were highly correlated within the SGs and that patients were more likely to have no STP or multiple STPs activated than expected (χ2 = 13.07, 3 degree of freedom [df], P = .001), and reconfirmed that STP activation was associated with a worse prognosis (Figure S3). Some described canonical pathway activities are not replicated in the dataset. For example, AKT is known to inactivate BAD and prevent apoptosis by phosphorylating serine 136 or to activate MTOR by phosphorylation. However, in this dataset when AKT.p473 and AKT.p308 is high, BAD.p136 and MTOR.p levels are low and vice versa, suggesting noncanonical interactions. Disconnects between canonical pathways and expression levels may provide clues to deregulated systems in leukemia cells. For example, in most SGs, high levels of PTEN.p are associated with low levels of phospho-AKT consistent with known models43 ; but in SG6, levels of AKT.p308 and .p473 are high despite high levels of pPTEN, suggesting a failure of PTEN to regulate phospho-AKT levels. This could represent a failed feedback loop where the cell up-regulated phospho-PTEN in an unsuccessful attempt to regulate phospho-AKT.43,44 Ratios of phophoprotein to total protein may provide additional insight.45 Detailed analysis of stored material from these cases, in combination with specific agents targeting these specific pathways, is required to test this hypothesis.
This dataset shows the power of being able to simultaneously ascertain the activation status of multiple pathways, with different functional effects, within a leukemic cell, a feature not possible with any prior method of analysis, including mRNA expression arrays. The combination of these patterns leads to the definition of protein expression SGs with prognostic significance for remission attainment, relapse, and survival. These SG classifications have prognostic significance that differs depending on the associated cytogenetics, suggesting that pathway activation must be considered in the context of the underlying genetics of the cell. Some cohorts, for example, unfavorable cytogenetics and SG6, have an unusually high CR rate and an unusually low relapse rate, suggesting that, despite having “poor prognosis cytogenetics,” patients with this signature do as well as favorable prognosis cytogenetics patients. A dismal outcome with current therapy can also be predicted in some groups, for example, unfavorable cytogenetics and SG1, with a 30% CR rate and a 100% relapse rate, suggesting that these patients should receive nontraditional therapies at presentation. Likewise, despite having favorable prognosis, cytogenetics patients with SG5 and SG6 had high relapse rates. Currently, stem cell transplantation for patients with favorable cytogenetics is reserved for those that relapse. These data, if confirmed, would suggest that favorable prognosis patients with these SGs should receive stem cell transplantation in first CR. Furthermore, in an era with a rapidly increasing number of targeted therapies, each with relatively narrow spectrum of activity, a means to rationally select which agent to use in what molecular background is both critical and unknown. The patterns of expression in these groups can be used to suggest rational combinations of targeted therapies that should be evaluated in combination with conventional therapies.
In conclusion, we were able to show the potential applicability of proteomic profiling for AML classification, target identification, treatment response prediction, and hypothesis generation for disease mechanisms. The complex pattern of intracellular protein expression and activation might harbor clues for disease pathobiology. It is possible to classify AML based on protein expression signatures that may predict outcome and therapy responsiveness across cytogenetics and other disease characteristics. Thus proteomic profiling may have advantages over other approaches of classifying AML as proteomic profiling may provide therapeutic guidance at the same time. A major question is whether the protein expression patterns defined from this dataset will be validated and found to be consistent across multiple patient sample sets, from multiple institutions, or have sufficient power to alter therapy. We are generating a validation array with a larger number of AML samples, which will be probed with an expanded number of antibodies to confirm our observation. If our results hold true, this would be a first step to the ultimate goal of personalized molecular therapy based on protein profiling.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This work was supported by the Leukemia & Lymphoma Society (grant 6089) and the National Institutes of Health (PO1 grant CA-55164).
National Institutes of Health
Authorship
Contribution: S.M.K. and R.T. designed research, performed research, analyzed data, and wrote the paper; Y.H.Q. performed research and analyzed data; W.C. generated the database and analyzed data; H.M.K. and M.A. performed clinical care and edited manuscript; K.R.C. performed statistical analysis and wrote the paper; and G.B.M. designed research, assisted with data analysis, and wrote the paper.
Conflict-of-interest disclosure: S.M.K., R.T., Y.H.Q., and K.R.C. have filed for a patent related to these discoveries. The remaining authors declare no competing financial interests.
Correspondence: Steven M. Kornblau, Section of Molecular Hematology and Therapy, Unit 448, University of Texas M. D. Anderson Cancer Center, 1515 Holcombe Blvd, Houston, TX 77030-4095; e-mail: skornblau@mdanderson.org.

{kind=link}
This feature is available to Subscribers Only
Sign In or Create an Account Close Modal