

Abstract
The sulfated polysaccharide fucoidan can rapidly mobilize hematopoietic progenitor cells (HPCs) and long-term repopulating stem cells from the bone marrow (BM) to the circulation. While searching for mechanisms involved in this phenomenon we found that BM myeloid cells bound to fucoidan through the integrin αMβ2 (macrophage antigen-1 [Mac-1]) and L-selectin resulting in αMβ2-independent release of neutrophil elastase, but inhibition of elastase activity did not impair fucoidan-induced mobilization. Mobilization of HPCs by fucoidan was enhanced in animals deficient in αM (αM–/–) compared with wild-type (αM+/+) animals and higher plasma levels of the chemokine CXCL12/stromal cell–derived factor-1 (SDF-1) were achieved in αM–/– mice by fucoidan treatment. However, in chimeric animals harboring αM+/+ and αM–/– HPCs in the BM, αM–/– HPCs were preferentially mobilized by fucoidan, suggesting that the enhanced mobilization is cell intrinsic and does not result from altered microenvironment. Suboptimal doses of granulocyte colony-stimulating factor (G-CSF) or cyclophosphamide (CY) also resulted in enhanced HPC mobilization in αM–/– mice compared with αM+/+ controls, but this difference was overcome when standard doses of G-CSF or CY were administered. Taken together, these data suggest that the integrin αMβ2 participates in the retention of HPCs in the BM.
Introduction
Billions of mature cells egress the bone marrow (BM) every day to contribute to the circulating blood cell elements. Under certain conditions, the egress of immature blood cells can be induced as well, a process referred to as mobilization. Several agents have been shown to induce hematopoietic progenitor cell (HPC) mobilization from the BM.1-3 However, the molecular mechanisms that regulate trafficking of HPCs between the blood and BM compartments are not fully understood. Emerging data indicate that HPC mobilization is a complex phenomenon involving the coordinated contributions of adhesion molecules, endogenous chemokines, proteases, and various signaling pathways that have not yet been identified. Mobilized circulating HPCs are clinically important as a source of hematopoietic stem cells in BM transplantation, a procedure that can provide a cure for certain cancers and benign hematologic disorders.
In contrast to the extravasation of leukocytes in which blood cells migrate from an environment subjected to shear stress to one that is relatively free of shear stress, mobilization of HPCs follows the opposite path. Thus, one would expect that the adhesion molecules that participate in cell anchorage or migration might play a more important role in HPC mobilization than those suited for shear-dependent interactions. Consistent with this idea, vascular cell adhesion molecule 1 (VCAM-1) has been proposed to retain HPCs in the BM since its blockade can increase circulating HPCs4 and it is proteolytically degraded during mobilization induced by granulocyte colony-stimulating factor (G-CSF) and cyclophosphamide (CY).5,6 In addition, G-CSF–induced mobilization is enhanced by antibodies against either major β2 integrins (αLβ2 or lymphocyte function-associated antigen 1 [LFA-1] and αMβ2), although mobilization was shown to proceed normally in the absence of αLβ2.7
The β2 integrin αMβ2 (also called CD11b/CD18 or macrophage antigen-1 [Mac-1]) is expressed on mature cells of the myeloid lineage and multipotent hematopoietic stem cells with limited self-renewal ability.8,9 αMβ2 displays highly promiscuous interactions with cellular and extracellular matrix ligands such as intercellular adhesion molecule-1 (ICAM-1) and -2, fibrinogen, iC3b, elastase, sulfated carbohydrates, zymosan, urokinase plasminogen activator, and β-glucan.10-17 In the context of transendothelial migration, αMβ2 binds ICAM-1 expressed on activated endothelium and mediates the arrest and diapedesis of leukocytes.18 Like most other integrins, the affinity of αMβ2 for ligands dramatically increases after activation by various stimuli, and its avidity is increased by the translocation to the cell surface of granular stores, a process that parallels the shedding of L-selectin from the surface of neutrophils.19,20 Although defects in leukocyte adhesion have been described in αM-deficient mice (αM–/–), these mice do not exhibit steady-state leukocytosis. In addition, αM–/– neutrophils exhibit deficient phagocytosis and reduced-oxygen free-radical generation and are more resistant to apoptosis.21
While evaluating the role of selectins in the mobilization process, we and others found that fucoidan, a sulfated fucose polymer known to interact with P- and L-selectins and to inhibit leukocyte rolling and inflammatory responses in vivo,22-24 can rapidly mobilize progenitors into the peripheral blood compartment.25,26 This effect was accentuated in endothelial selectin–deficient mice, indicating that the presence of selectins was not required for fucoidan-induced mobilization. Further studies have suggested that fucoidan increased the levels of stromal cell–derived factor (SDF-1 or CXCL12) in the blood, an effect that may contribute to its mobilizing activity.27 Since mobilization activity has also been linked to myeloid cells,6,28-30 we evaluated their interactions with fucoidan. Here, we show that αMβ2 and L-selectin are the major receptors for fucoidan on myeloid cells. Fucoidan binding produced the release of neutrophil proteases without evidence of classical myeloid cell activation. However, neither the absence of αMβ2 nor elastase inhibition impaired fucoidan-induced HPC mobilization. HPC mobilization was in fact increased in the absence of αMβ2 whether induced by fucoidan, suboptimal doses of G-CSF or CY. Our results suggest that αMβ2 contributes to anchor HPCs in the BM parenchyma.
Materials and methods
Animals
L-selectin–deficient mice (L–/–; from Dr Richard Hynes, Massachusetts Institute of Technology, Cambridge, MA)31 and αM–/– mice (from Dr Tanya Mayadas, Harvard Medical School, Boston, MA)21 were generated by homologous recombination. Breeding stocks of the 2 knockout strains and their wild-type counterparts were of the same C57BL/6/129Sv mixed background. C3/HeJ mice were obtained from Jackson Laboratories (Bar Harbor, ME), and C57/BL6 were obtained from the National Cancer Institute (Frederick Cancer Research and Developmental Center, Frederick, MD). Transgenic mice expressing enhanced green fluorescence protein (EGFP) under the control of the β-actin promoter were from Dr Sergio Lira (Mount Sinai School of Medicine). All mice used for experiments were age- and sex-matched (6-9 weeks). Mice were bred and housed at Mount Sinai School of Medicine animal facilities. Experimental procedures performed on the animals were approved by the Animal Care and Use Committee of Mount Sinai.
Antibodies and preparation of reagents
Rat immunoglobulin G (IgG; Sigma, St Louis, MO) was reconstituted in phosphate-buffered saline (PBS). Rat anti–L-selectin (clone MEL-14; IgG2a;American Type Culture Collection [ATCC], Rockville, MD) and rat anti-αM (clone M1/70; IgG2b; ATCC) were purified from supernatants using a protein G column (HiTrap Protein G Sepharose HP; Amersham Pharmacia, Uppsala, Sweden). Potential endotoxin contamination in the rat IgG was removed by running the suspension through a polymixin B column (Detoxi-Gel; Pierce, Rockford, IL). Antibodies against TER119 and B220 were purchased from BD Pharmingen (San Diego, CA).
For fucoidan biotinylation, fucoidan (Fluka, lot no. 385468/1; Chemie AG, Buchs, Switzerland) was dissolved in endotoxin-free PBS. Fucoidan was oxidized by sodium periodate treatment25 to create reactive aldehyde groups that will form a hydrazone linkage with EZ-Link Biotin-LC-Hydrazide (Pierce). The concentration of biotinylated fucoidan (B-Fuc) was measured according to the method of Dubois et al32 using native fucoidan (N-Fuc) as standard.
Bone marrow cell isolation, labeling, and activation
Bone marrow nucleated cells (BMNCs) were harvested by flushing femora in RPMI using a 21-gauge needle. A single-cell suspension was obtained by gently passing the flushed marrow through the needle several times. Contaminating erythrocytes (red blood cells [RBCs]) were lysed in 0.8% NH4Cl and the remaining leukocytes (white blood cells [WBCs]) were washed twice in RPMI containing 5% fetal bovine serum (FBS). Cells (5 × 105/100 μL/sample) were incubated in RPMI plus 5% FBS in the presence of saturating concentrations of B-Fuc (150 μg/mL in 100 μL) and stained with fluorescein isothiocyanate (FITC)–conjugated streptavidin (Jackson ImmunoResearch, West Grove, PA). Data were collected by flow cytometry from gated myeloid cells (high side-scatter fraction of panleukocyte gate). In control samples, cells were pretreated with 200 μg N-Fuc, EDTA (ethylenediaminetetraacetic acid; 5 mM), or vehicle for 30 minutes at 4° C.
To determine the activation state of myeloid cells, surface expression of L-selectin and αMβ2 and oxidative burst were measured by flow cytometry. Total BMNCs (5 × 105) were incubated with N-Fuc (100 μg/mL), phorbol-myristate acetate (PMA; Sigma; 50 ng/mL) or vehicle in Tris-buffered saline (TBS) containing 1 mM CaCl2 and 0.5 mM MgCl2 for 30 minutes at 37° C. Cells were then incubated with antibodies against L-selectin or αM (10 μg/mL) for 15 minutes at 6° C, washed with cold PBS, and further incubated with a cyanin 5 (Cy5)–labeled antirat antibody before a final wash. To measure the oxidative burst, 5 × 105 cells were washed and resuspended in 300 μL of PBS containing 5μM dichlorofluorescein diacetate (DCFDA; Molecular Probes, Eugene, OR). After an additional incubation in PBS, analysis was performed on a FACSCalibur flow cytometer (Becton Dickinson, Mountain View, CA). Unless otherwise indicated, all incubations were performed for 15 minutes at room temperature (RT).
Static adhesion assays
BMNCs were harvested and fluorescently labeled with 33 μM CFSE (5-(and-6)-carboxyfluorescein diacetate, succinimidyl ester; Molecular Probes) by 20 minutes incubation at RT. BMNCs were preincubated with antibodies (10 μg/mL; MEL14, M1/70, or both) or sugar (1 mg/mL; fucoidan, mannan, heparin, glucose, and fucose, all from Sigma) for 10 minutes at RT. Cells (7 × 104/well) were layered on hydrazide-coated 96-well plates (Carbo-Bind; Corning-Costar, Acton, MA) that had been precoated with 100 μL of activated fucoidan at the indicated concentrations, followed by blocking (0.5% bovine serum albumin [BSA] in PBS for 2 hours at RT), and then cells were allowed to adhere for 30 minutes at 37° C. Wells were washed thrice with RPMI, bound cells were lysed with 0.1% sodium dodecyl sulfate (SDS) in PBS, and fluorescence was read with a cytofluorometer (485/535 nm). In the experiments where binding of culture colony-forming units (CFU-Cs) was assayed, unlabeled bound BMNCs were detached by incubation with trypsin for 5 minutes and plated in methylcellulose cultures for CFU-C content.25 In some experiments, detached cells were analyzed by flow cytometry for the expression of markers for the erythroid, lymphoid, and myeloid lineages using the TER119, B220, and M1/70 antibodies, respectively.
Fucoidan-induced enzymatic release
BMNCs were prepared from αM+/+, αM–/–, or C3/HeJ mice and resuspended (2 × 106 cells/mL) in assay buffer (Tris 50 mM, pH 7.5; 150 mM NaCl; 1 mM CaCl2; 0.5 mM MgCl2). Cells were incubated for 30 minutes at 37° C with 50 ng/mL PMA, 100 μg/mL fucoidan, or vehicle. Cells were then centrifuged and supernatant was collected using the Handee Resin Separators (Pierce) to prevent contamination of the supernatants with fucoidan-induced neutrophil aggregates. To assess elastase activity, 60 μL of supernatants was incubated with an equal volume of a buffer containing 0.1% Brij 35, 0.5 M NaCl, and 1 mM elastase substrate I (Calbiochem, La Jolla, CA). The reaction was allowed to proceed for at least 1 hour at 37° C before optical density (OD) was measured at 405 nm.
Progenitor mobilization
Fucoidan-induced progenitor mobilization was performed in αM+/+ and αM–/– mice. Animals were injected twice intraperitoneally with 50 mg/kg fucoidan 4 hours apart, and blood and bone marrow were harvested 2 hours after the second dose. Blood was obtained by retro-orbital sampling of mice anesthetized with tribromoethanol and collected in tubes containing EDTA. Blood counts were obtained using an automated cell counter (Serono-Baker Diagnostics, Allentown, PA). Mononuclear cells were isolated and plated for CFU-C determination. To test the role of elastase in fucoidan-induced mobilization, the elastase inhibitor III (MeOSuc-AAPV-CMK; EMD Biosciences, San Diego, CA) or vehicle control were injected intraperitoneally (1 mg per mouse) 2 hours prior to the first dose of fucoidan.
Mobilization by CY was achieved as described by Liu et al.29 αM+/+ and αM–/– mice were injected intraperitoneally with a single dose of 200 or 100 mg/kg of CY (Sigma). Blood and bone marrow were harvested 8 days after CY administration and plated for CFU-C assay.
For G-CSF–induced mobilization, αM+/+ and αM–/– mice were treated twice daily with filgrastim (250 μg/kg/d for 1, 2, or 4 days by subcutaneous injection; Amgen, Thousand Oaks, CA) diluted in endotoxin-free PBS containing 0.1% BSA. Three hours after the final injection, peripheral blood and BMNCs were harvested and plated in methylcellulose as described above for the static adhesion assays.
Competitive mobilization experiments
We used a competitive mobilization assay25 to compare the ability of αM+/+ and αM–/– progenitors to be mobilized after fucoidan treatment in the same mouse. Mice chimeric for αMβ2 and EGFP expression were generated by BM transplantation. Eight-week-old C57/BL6 animals were used as recipients and were lethally irradiated (12 Gy; 2 split doses, 3 hours apart) from a cesium source (Model I-68A; J. L. Shepherd, San Fernando, CA). Irradiated mice were then injected via the lateral tail vein with a mixture of BMNCs obtained from αM–/– or αM+/+ mice combined with BMNCs from EGFP transgenic mice, which express wild-type levels of αM (1.5 × 106 EGFP BMNCs mixed with 1.5 × 106 nonfluorescent BMNCs either αM+/+ or αM–/–). Two months after transplantation, reconstitution of hematopoiesis was assessed by flow cytometry to detect EGFP-positive cells and αM expression (by staining with phycoerythrin [PE]–conjugated anti-αM antibody [Ab]; clone M1/70; eBioscience, San Diego, CA). Expression was found to be approximately in a 1:1 ratio for GFP-positive and GFP-negative cells in the peripheral blood for both groups of animals. Chimeric mice were injected with a single dose of fucoidan (100 mg/kg), which induces mobilization similar to that of the 2-dose schedule (not shown), or saline control, and blood was harvested 2 hours after treatment to assess circulating CFU-Cs. Nonfluorescent and green fluorescent colonies were identified by light and fluorescence microscopy, using an Axiovert S100 TV inverted microscope (Zeiss, Göttingen, Germany) with a × 5 dry objective (0.25 NA). Images were captured with a Spot Insight Color camera and analyzed with Spot 3.2.2 software (both from Diagnostic Instruments, Sterling Heights, MI).
CXCL12 ELISA
BM samples were prepared by flushing 2 femurs with 1 mL of cold PBS. Cells were centrifuged (300g) to collect supernatant. Peripheral blood plasma was collected by centrifugation of blood at 8000g and 4° C. Samples were stored at –80° C until use and then diluted 2 and 5 times in PBS for analysis. The levels of CXCL12 were measured by enzyme-linked immunosorbent assay (ELISA; R&D Systems, Minneapolis, MN) exactly as described by Petit et al.30
Chemotactic assays
BMNCs were incubated with the indicated concentrations of fucoidan for 30 minutes at 37° C and then transferred to the upper chamber of Transwells (5-μM pore size; Corning-Costar) in 100 μL of assay buffer (RPMI containing 0.5% BSA). In the indicated samples, cells were washed once before transferring to the Transwells to remove unbound fucoidan. Migration was allowed to proceed for 1 hour at 37° C toward a concentration of 25 ng/mL CXCL12 (R&D Systems). Migrated and input cells were assayed for CFU-Cs as described.25 The percentage of migrated CFU-Cs was calculated from the input number for each sample.
Statistical analysis
All values are reported as a mean ± SEM. Statistical significance for 2 unpaired groups was assessed by the Student t test.
Results
Fucoidan binds to nonselectin receptors on myeloid cells
Since fucoidan can mobilize progenitors from the BM to the blood compartment in a selectin-independent manner25,26 and recent experimental evidence has linked mobilization with myeloid cells,2,5,6,29 we hypothesized that fucoidan might interact with other receptors on the surface of myeloid cells. To investigate this possibility, BMNCs were stained with biotinylated fucoidan (B-Fuc) and bound fucoidan detected by flow cytometry. As shown in Figure 1, B-Fuc bound to virtually all BM myeloid cells. Binding was partially blocked by 5 mM EDTA, suggesting that divalent cations were in part required. B-Fuc binding was specific since preincubation with native fucoidan (N-Fuc) effectively competed with B-Fuc binding. The residual B-Fuc binding after N-Fuc preincubation may result from the generation of nonspecific binding motifs by the biotinylation reaction. Interestingly, B-Fuc staining was only partially abrogated in myeloid cells lacking L-selectin (Figure 1; Table 1), suggesting that fucoidan can interact with nonselectin receptor(s).

Fucoidan specifically binds bone marrow myeloid cells via L-selectin and other receptors. BMNCs from wild-type mice were incubated with biotinylated fucoidan (B-Fuc, filled histogram), native fucoidan (N-Fuc, bold line histogram), followed by B-Fuc, or B-Fuc in the presence of EDTA (dashed-line histogram). Also shown is the staining with B-Fuc of BMNCs from L–/– mice (thin-line histogram).
Fucoidan specifically binds bone marrow myeloid cells via L-selectin and other receptors. BMNCs from wild-type mice were incubated with biotinylated fucoidan (B-Fuc, filled histogram), native fucoidan (N-Fuc, bold line histogram), followed by B-Fuc, or B-Fuc in the presence of EDTA (dashed-line histogram). Also shown is the staining with B-Fuc of BMNCs from L–/– mice (thin-line histogram).
Binding of biotinylated fucoidan to BMNCs
Genotype | B-Fuc | EDTA + B-Fuc | N-Fuc + B-Fuc |
|---|---|---|---|
| WT | 54 ± 2 | 24 ± 2 | 16 ± 3 |
| αM-/- | 35 ± 5* | 21 ± 3 | 13 ± 2 |
| L-/- | 30 ± 2* | 25 ± 1 | 17 ± 2 |
Genotype | B-Fuc | EDTA + B-Fuc | N-Fuc + B-Fuc |
|---|---|---|---|
| WT | 54 ± 2 | 24 ± 2 | 16 ± 3 |
| αM-/- | 35 ± 5* | 21 ± 3 | 13 ± 2 |
| L-/- | 30 ± 2* | 25 ± 1 | 17 ± 2 |
BMNCs were isolated from WT, L-/-, and αM-/- mice and labeled with 15 μg of biotinylated fucoidan (B-Fuc) alone or in the presence of 5 mM EDTA or after preincubation with 200 μg native fucoidan (N-Fuc). Geometric means of fluorescence ± SEM were determined by flow cytometric analysis following secondary labeling with FITC-labeled streptavidin.
WT indicates wild type.
P < .05; n = 3-7 mice.
Bone marrow nucleated cells bind fucoidan via L-selectin and the integrin αMβ2
To further characterize the receptors involved in fucoidan binding, we set up an in vitro static adhesion assay. Hydrazide plates were coated with a range of concentrations of fucoidan, desulfated fucoidan, or heparin. Fluorescently labeled cells were incubated in sugar-coated wells and the number of bound cells was assessed by measuring the fluorescence remaining in the wells. As shown in Figure 2A, cells bound to immobilized fucoidan in a dose-dependant manner, reaching a plateau at a sugar concentration of approximately 1 mg/mL. This fucoidan concentration was thus used in subsequent static adhesion experiments. Adhesion of BMNCs was significantly reduced when plates were coated with desulfated fucoidan. Comparatively, very few cells bound to immobilized heparin (Figure 2A). To further investigate the specificity of binding to fucoidan, we preincubated cells with various sugars including fucoidan, mannan, heparin, glucose, and fucose. We found that soluble fucoidan, but not the other carbohydrates, inhibited cell binding to immobilized fucoidan (Figure 2B). To assess whether fucoidan bound to specific subsets of BM mature or precursor cells, we evaluated the lineage of fucoidan-binding cells by flow cytometry. We found that bound cells were significantly enriched in myeloid cells (Mac-1+), whereas significantly fewer erythroid precursors (TER119+) interacted with fucoidan (Table 2). Furthermore, approximately 30% of plated CFU-Cs bound to immobilized fucoidan and this binding could be completely inhibited by soluble fucoidan (Figure 2C).

BMNCs bind to immobilized fucoidan through L-selectin and αMβ2. (A) Dose-response binding. CFSE-labeled BMNCs were allowed to bind to hydrazide-coated wells that had been precoated with various concentrations of fucoidan (•), desulfated-fucoidan (D-Fuc; □), or heparin (▴). The wells were then washed and cells were lysed. Fluorescence was measured and the number of bound BMNCs was calculated. (B) Binding specificity of fucoidan. CFSE-labeled BMNCs were allowed to bind to plates coated with fucoidan in the presence of EDTA or sugars (fucoidan, mannan, heparin, glucose, or fucose); *P ≤ .009 compared with the control (Nil) group. (C) Progenitor binding to fucoidan. BMNCs preincubated with fucoidan (N-Fuc) or media (Nil) were allowed to bind BSA (▨) or immobilized fucoidan. Bound CFU-Cs were scored in methylcellulose cultures. *P < .05 compared with BSA and N-Fuc. Binding in the untreated group (Nil) corresponds to 29% ± 2% of the input CFU-Cs. (D) BMNCs binding to immobilized fucoidan are mediated by L-selectin and the αMβ2 integrin. CFSE-labeled BMNCs were allowed to bind to fucoidan-coated plates in the presence of antirat IgG control, anti–L-selectin antibody (MEL14), anti-αM antibody (M1/70), or in the presence of both function-blocking antibodies. In all panels, error bars represent mean ± SEM bound cells/mm2. *P < .05; **P ≤ .01 compared with vehicle (Nil) or control IgG groups.
BMNCs bind to immobilized fucoidan through L-selectin and αMβ2. (A) Dose-response binding. CFSE-labeled BMNCs were allowed to bind to hydrazide-coated wells that had been precoated with various concentrations of fucoidan (•), desulfated-fucoidan (D-Fuc; □), or heparin (▴). The wells were then washed and cells were lysed. Fluorescence was measured and the number of bound BMNCs was calculated. (B) Binding specificity of fucoidan. CFSE-labeled BMNCs were allowed to bind to plates coated with fucoidan in the presence of EDTA or sugars (fucoidan, mannan, heparin, glucose, or fucose); *P ≤ .009 compared with the control (Nil) group. (C) Progenitor binding to fucoidan. BMNCs preincubated with fucoidan (N-Fuc) or media (Nil) were allowed to bind BSA (▨) or immobilized fucoidan. Bound CFU-Cs were scored in methylcellulose cultures. *P < .05 compared with BSA and N-Fuc. Binding in the untreated group (Nil) corresponds to 29% ± 2% of the input CFU-Cs. (D) BMNCs binding to immobilized fucoidan are mediated by L-selectin and the αMβ2 integrin. CFSE-labeled BMNCs were allowed to bind to fucoidan-coated plates in the presence of antirat IgG control, anti–L-selectin antibody (MEL14), anti-αM antibody (M1/70), or in the presence of both function-blocking antibodies. In all panels, error bars represent mean ± SEM bound cells/mm2. *P < .05; **P ≤ .01 compared with vehicle (Nil) or control IgG groups.
Differential binding of BMNCs to fucoidan
Cell type | Input, % | Bound, % | P |
|---|---|---|---|
| Erythroid, TER119+ | 12.3 ± 1.2 | 2.2 ± 0.5 | .0004 |
| B-lymphoid, B220+ | 34.3 ± 3.2 | 28.2 ± 1.6 | .1305 |
| Myeloid, Mac-1+ | 52.1 ± 0.8 | 72.9 ± 0.6 | .0045 |
Cell type | Input, % | Bound, % | P |
|---|---|---|---|
| Erythroid, TER119+ | 12.3 ± 1.2 | 2.2 ± 0.5 | .0004 |
| B-lymphoid, B220+ | 34.3 ± 3.2 | 28.2 ± 1.6 | .1305 |
| Myeloid, Mac-1+ | 52.1 ± 0.8 | 72.9 ± 0.6 | .0045 |
BMNCs from wild-type mice were allowed to adhere to immobilized fucoidan for 30 minutes. Bound cells were detached with trypsin/EDTA and stained with the antibodies TER119, B220, and M1/70 to stain for cells of the erythroid, lymphoid, and myeloid lineages, respectively. Input BMNCs used for the experiment were also stained for comparison. Data are expressed as mean percentages ± SEM from 3 to 5 mice in 3 independent experiments. Note that only the myeloid population is significantly enriched in the bound population while the erythroid population is depleted.
In addition to the known interactions with L-selectin,33 previous studies have shown that negatively charged carbohydrates can interact with activated13,34,35 or purified17 αMβ2. To test their role in the adhesion to immobilized fucoidan, fluorescently labeled BMNCs were preincubated with anti–L-selectin (MEL14), anti-αM (M1/70), both, or control rat IgG before being plated on hydrazide plates coated with fucoidan. Fluorescence of bound cells was measured after cell lysis. As shown in Figure 2D, BMNC binding to fucoidan was mildly inhibited by the anti–L-selectin (∼15%) but greatly blocked by the anti-αM (∼60%) compared with control IgG. Consistent with these results, binding to fucoidan of L–/– BMNCs was slightly lower than that of wild-type cells (∼20% reduction in control IgG-treated L–/– cells), but this difference was not statistically significant. Binding to fucoidan was maximally reduced (∼75%) when antibodies to both L-selectin and αM were used simultaneously in wild-type cells (by an additional ∼15% compared with only αM inhibition) or when αM was blocked on cells from L–/– mice (Figure 2D).
To further evaluate the specificity of fucoidan interactions, we compared the ability of soluble biotinylated fucoidan to bind to BMNCs obtained from mice genetically deficient in αMβ2 or L-selectin and wild-type mice. As shown in Table 1, B-Fuc binding to L–/– BMNCs was significantly reduced compared with that of wild-type animals. Binding to αM–/– cells was also significantly reduced, but in contrast to the above static adhesion assay on immobilized fucoidan, the reduced binding in this fluid-phase assay was greater for L–/– cells than for αM–/– cells (37% and 28% reduction, respectively; Table 1). The simultaneous contributions of both L-selectin and αMβ2 integrin could not be assessed in this assay because control IgG itself altered the fluorescent readout (data not shown). Thus, these results indicate that fucoidan, both in soluble or immobilized forms, can interact with the αMβ2 integrin and L-selectin and that this interaction does not require prior myeloid cell activation.
Fucoidan induces the release of serine proteases from myeloid cells
αMβ2-mediated binding without exogenous cell activation suggested that fucoidan can either bind unactivated αMβ2 or that fucoidan might directly activate myeloid cells. One hallmark of myeloid cell activation is the release of granular serine proteases, such as elastase and cathepsin G. In particular, elastase has recently been suggested to play a critical role in HPC mobilization through the degradation of VCAM-1, CXCL12, and CXCR4.5,30,36 To assess whether fucoidan can induce the release of elastase, BMNCs were incubated in the presence of fucoidan, and of vehicle, or PMA as negative and positive controls, respectively. Protease activities released in the supernatant were then measured using a chromogenic substrate. Dose-response experiments revealed a plateau in the elastase activity release at concentrations of fucoidan of at least 100 μg/mL (not shown). As depicted in Figure 3A, fucoidan induced a significant increase in the levels of elastase activity in the extracellular milieu (4.5-fold increase). Since αMβ2 can interact with both fucoidan and elastase,11 we also analyzed the ability of αM–/– cells to release elastase. αM–/– cells treated with fucoidan, however, released levels of elastase similar to those of control αM+/+ BMNCs (2.7-fold increase; P = .46 compared with αM+/+). To rule out the possibility that trace endotoxin contamination may have induced neutrophil activation and release of these proteases, we repeated the above enzymatic assays on BMNCs isolated from C3H/HeJ mice, which are unresponsive to endotoxin due to a mutation in the Tlr4 gene.37 We found that fucoidan also induced Tlr4-deficient myeloid cells to release elastase (Figure 3A). Interestingly, fucoidan-induced release of elastase from BMNCs was stronger than that of 50 ng/mL PMA (4.5-fold vs 1.7-fold increase, respectively; P = .02). Similar results were obtained for cathepsin G release, using a substrate specific for this protease (data not shown). Other experiments performed using purified BM neutrophils also yielded similar results (not shown), indicating that neutrophils were the main source of released serine proteases.

Fucoidan induces the release of proteases without activation of myeloid cells. (A) BMNCs from αM+/+, αM–/–, or C3H/HeJ mice were treated with 100 μg/mL fucoidan, vehicle, or with 50 ng/mL PMA (C3H/HeJ group only). Enzymatic activities were measured using a chromogenic substrate. Error bars represent mean ± SEM relative elastase activity. *P < .01; **P < .05, n = 4. (B) Expression of L-selectin or αM in vehicle, fucoidan, and PMA-treated BMNCs and (C) the oxidative burst as measured with DCFDA were analyzed by flow cytometry. Vertical lines indicate the fluorescence level of untreated or negative control samples. Shown are representative of at least 3 experiments.
Fucoidan induces the release of proteases without activation of myeloid cells. (A) BMNCs from αM+/+, αM–/–, or C3H/HeJ mice were treated with 100 μg/mL fucoidan, vehicle, or with 50 ng/mL PMA (C3H/HeJ group only). Enzymatic activities were measured using a chromogenic substrate. Error bars represent mean ± SEM relative elastase activity. *P < .01; **P < .05, n = 4. (B) Expression of L-selectin or αM in vehicle, fucoidan, and PMA-treated BMNCs and (C) the oxidative burst as measured with DCFDA were analyzed by flow cytometry. Vertical lines indicate the fluorescence level of untreated or negative control samples. Shown are representative of at least 3 experiments.
To determine whether the release of these proteases was due to neutrophil activation by fucoidan, fucoidan- or PMA-treated BMNCs were evaluated for expression of L-selectin and αMβ2, which are down-regulated and up-regulated, respectively, upon activation. In contrast to PMA, fucoidan treatment did not alter the expression of L-selectin or αM on neutrophils (Figure 3B). In addition, fucoidan was unable to induce an oxidative burst in BM neutrophils as assessed by cytometric analysis using the DCFDA probe21 (Figure 3C). These results indicate fucoidan does not appear to induce bona fide myeloid cell activation and that serine proteases may be released from the cell surface11 in a manner that does not require αMβ2 integrin.
To determine whether the release of elastase induced by fucoidan might be related to its capacity to trigger mobilization, mice were injected with an elastase inhibitor 2 hours prior to the first fucoidan injection. In contrast with G-CSF–induced mobilization,30 elastase inhibition did not reduce mobilization by fucoidan (291 ± 66 CFU-Cs/mL of blood in control vehicle-treated mice vs 360 ± 85 in inhibitor-treated mice, mean ± SEM; n = 10 mice per group; P = .57). These results indicate that fucoidan-induced mobilization is independent of the release of elastase activity.
Fucoidan-induced mobilization is enhanced in the absence of the integrin αMβ2
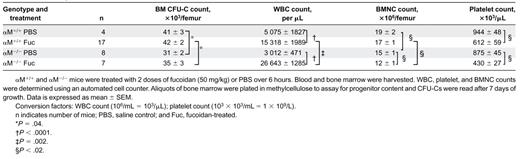
To evaluate whether the expression of αMβ2influenced the ability of fucoidan to mobilize HPCs from the BM, we treated αM–/– and wild-type control mice with 2 doses of 50 mg/kg fucoidan over a 6-hour period. Two hours after the last injection, peripheral blood and BM were harvested, counts were determined, and nucleated cells were assayed for CFU-Cs. As shown in Figure 4, there was a 5-fold increase in HPC mobilization in fucoidan-treated αM+/+ mice. HPC mobilization was further increased in fucoidan-treated αM–/– mice (24.5-fold higher than in vehicle-treated animals; P < .0001; and 3.7-fold higher than fucoidan-treated wild-type animals; P < .0001). Fucoidan-treated mice from both genotypes exhibited similar leukocytosis, thrombocytopenia, and reduced BMNCs compared with their vehicle-treated counterparts (Table 3). The increased HPC mobilization in αM–/– mice occurred despite fewer resident BM CFU-Cs in these mice compared with αM+/+ animals (Table 3).

Enhanced mobilization in αM–/– mice by fucoidan. αM+/+ and αM–/– mice were treated with 2 doses of fucoidan (50 mg/kg each; ▪) or PBS (□). Nucleated cells isolated from peripheral blood 6 hours after treatment were assayed for CFU-C content. Error bars represent mean ± SEM values. *P < .005; n = 5-24.
Enhanced mobilization in αM–/– mice by fucoidan. αM+/+ and αM–/– mice were treated with 2 doses of fucoidan (50 mg/kg each; ▪) or PBS (□). Nucleated cells isolated from peripheral blood 6 hours after treatment were assayed for CFU-C content. Error bars represent mean ± SEM values. *P < .005; n = 5-24.
Higher plasma levels of CXCL12 in fucoidan-treated αM–/– mice
Recent reports have shown a correlation between mobilization and degradation of the chemokine CXCL12 in the BM of G-CSF–mobilized mice.30,36 In addition, mice mobilized with fucoidan display increased levels of CXCL12 in peripheral blood plasma and reduced levels in the BM.27 To investigate whether the enhanced mobilization of HPCs in αM–/– mice was related to the levels of CXCL12 in BM and blood, wild-type or αM–/– mice were mobilized with fucoidan and CXCL12 levels were quantitated by ELISA. While BM CXCL12 levels of αM+/+ and αM–/– mice did not differ significantly at baseline and were similarly reduced after fucoidan treatment (Figure 5A), plasma levels of CXCL12 were significantly higher in αM–/– mice following fucoidan treatment (Figure 5B).

Increased levels of CXCL12 in the plasma of αM–/– mice after fucoidan mobilization. αM+/+ and αM–/– mice were treated with 2 doses of PBS (□) or fucoidan (50 mg/kg; ▪). BM extracellular fluid (A) and plasma (B) were harvested for analysis of CXCL12 content by ELISA. Shown is the mean ± SEM concentration of CXCL12 from 3 independent experiments. *P < .01; **P < .0001; #P = .02 compared with PBS; ¥P = .04. n = 6-12 mice.
Increased levels of CXCL12 in the plasma of αM–/– mice after fucoidan mobilization. αM+/+ and αM–/– mice were treated with 2 doses of PBS (□) or fucoidan (50 mg/kg; ▪). BM extracellular fluid (A) and plasma (B) were harvested for analysis of CXCL12 content by ELISA. Shown is the mean ± SEM concentration of CXCL12 from 3 independent experiments. *P < .01; **P < .0001; #P = .02 compared with PBS; ¥P = .04. n = 6-12 mice.
Soluble fucoidan enhances the migration of HPCs toward CXCL12
It has recently been shown that cross-linking of surface L-selectin by fucoidan can up-regulate the expression and function of CXCR4 on human lymphocytes.38 To assess whether this was the case for lineageneg Sca-1+ c-Kit+ cells, we incubated BMNCs with fucoidan and assessed CXCR4 expression by flow cytometry. We found that fucoidan did not up-regulate CXCR4 expression on lineageneg Sca-1+ c-Kit+ cells (not shown). We next analyzed the effect of fucoidan in the capacity of HPCs to migrate toward CXCL12 in vitro. Suboptimal concentrations of CXCL12 (25 ng/mL) were used because they are similar to those found in the plasma of fucoidan-injected mice (Figure 5B) and render the assay more sensitive. We found that relatively low concentrations (10 μg/mL) of fucoidan significantly and reproducibly enhanced progenitor migration, whereas higher concentrations (100 μg/mL) were inhibitory (Figure 6). To differentiate the possibility of fucoidan-induced cell signaling from a possible interaction of soluble fucoidan with CXCL12,27 we removed fucoidan by washing cells prior to the migration assay. Interestingly, the enhanced migration of CFU-Cs was abrogated when unbound fucoidan was removed (Figure 6). This suggests that soluble fucoidan may enhance CFU-C migration by enabling a more efficient presentation of the chemokine to HPCs rather than by triggering intracellular signals within the cells.

Fucoidan alters CFU-C migration toward CXCL12. BMNCs were preincubated with different concentrations of fucoidan before performing chemotactic assays to 25 ng/mL CXCL12. Migrated and input cells were assayed for CFU-Cs in methylcellulose cultures. In some samples, cells were washed before the migration assay to remove unbound fucoidan (10+wash). Shown is the mean ± SEM of migrated CFU-Cs. n = 3-5 independent experiments; ¥ P < .005. One hundred percent migration reflects 7.3% ± 1.8% of input CFU-Cs.
Fucoidan alters CFU-C migration toward CXCL12. BMNCs were preincubated with different concentrations of fucoidan before performing chemotactic assays to 25 ng/mL CXCL12. Migrated and input cells were assayed for CFU-Cs in methylcellulose cultures. In some samples, cells were washed before the migration assay to remove unbound fucoidan (10+wash). Shown is the mean ± SEM of migrated CFU-Cs. n = 3-5 independent experiments; ¥ P < .005. One hundred percent migration reflects 7.3% ± 1.8% of input CFU-Cs.
Enhanced competitive mobilization of αM–/– HPCs by fucoidan
To distinguish between the possibility that the enhanced mobilization in αM–/– mice results from an altered microenvironment and the possibility of cell intrinsic abnormality, we established a competitive mobilization assay. Lethally irradiated C57/BL6 recipient mice were reconstituted with a mixture of BM cells from αMβ2-sufficient EGFP-transgenic mice and EGFP-negative BM cells from either αM-sufficient or αM-deficient mice. Two months after transplantation, both groups of chimeric mice (αM+/+ EGFP-negative with αM+/+ EGFP-positive cells and αM–/– EGFP-negative with αM+/+ EGFP-positive cells) presented near 50% of circulating EGFP-positive cells by flow cytometry and a similar ratio of EGFP-negative/EGFP-positive CFU-Cs in the BM, suggesting a similar engraftment of both groups (not shown). Mice were then mobilized with fucoidan and the relative numbers of nonfluorescent and EGFP-positive colonies were assessed using light and fluorescence microscopy (Figure 7B; Table 4). In the control group in which both nonfluorescent and fluorescent HPCs expressed αM, their ratio was close to 1, suggesting that EGFP-negative and EGFP-positive cells are mobilized equally well by fucoidan. However, in the group harboring αM–/– and αM+/+ EGFP-positive cells, αM–/– HPCs were predominantly mobilized (Figure 7A; Table 4). These data suggest that the enhanced release of progenitors by fucoidan in αM–/– mice is intrinsic to the cell itself and may not result from an altered BM microenvironment.

Progenitor mobilization in chimeric mice. Animals chimeric for αMβ2 (αM+/+ or αM–/–) and enhanced green fluorescence protein (EGFP) expression were generated by bone marrow transplantation. Two months after transplantation mice were mobilized by a single injection of fucoidan. The numbers of nonfluorescent and green fluorescent colonies after 7 days of culture were determined using an inverted microscope equipped with a fluorescence source. (A) The ratios of EGFP-negative and EGFP-positive colonies were determined from mobilized colonies in the blood. Shown are mean ± SEM ratios. n = 6 from 2 independent experiments (Table 3). *P < .05 compared with the EGFP-αM+/+ chimera. (B) A representative area containing colonies grown in methylcellulose media is shown using phase (top) or fluorescence microscopy (bottom) and a × 5 dry objective. EGFP-derived HPCs (arrowheads) can be distinguished from nonfluorescent colonies (arrows).
Progenitor mobilization in chimeric mice. Animals chimeric for αMβ2 (αM+/+ or αM–/–) and enhanced green fluorescence protein (EGFP) expression were generated by bone marrow transplantation. Two months after transplantation mice were mobilized by a single injection of fucoidan. The numbers of nonfluorescent and green fluorescent colonies after 7 days of culture were determined using an inverted microscope equipped with a fluorescence source. (A) The ratios of EGFP-negative and EGFP-positive colonies were determined from mobilized colonies in the blood. Shown are mean ± SEM ratios. n = 6 from 2 independent experiments (Table 3). *P < .05 compared with the EGFP-αM+/+ chimera. (B) A representative area containing colonies grown in methylcellulose media is shown using phase (top) or fluorescence microscopy (bottom) and a × 5 dry objective. EGFP-derived HPCs (arrowheads) can be distinguished from nonfluorescent colonies (arrows).
Cell and progenitor counts after fucoidan mobilization in EGFP-αM+/+ and EGFP-αM–/– chimeric mice
Genotype and treatment | WBCs, × 106/mL blood | CFUs/mL of blood | |||||
|---|---|---|---|---|---|---|---|
| n of mice | EGFP- | EGFP+ | Ratio | ||||
| Experiment 1 | |||||||
| αM+/+ | |||||||
| PBS | 2 | 3.9 ± 0.1 | 0 ± 0 | 0 ± 0 | NA | ||
| Fuc | 3 | 31.7 ± 6 | 283 ± 18 | 270 ± 32 | 1.04 | ||
| αM-/- | |||||||
| PBS | 2 | 5.0 ± 0.8 | 7 ± 3 | 0 ± 0 | NA | ||
| Fuc | 3 | 25.2 ± 2 | 280 ± 55 | 147 ± 29 | 1.90 | ||
| Experiment 2 | |||||||
| αM+/+ | |||||||
| PBS | 2 | 2.6 ± 0.0 | 0 ± 0 | 0 ± 0 | NA | ||
| Fuc | 3 | 18.2 ± 3.2 | 207 ± 48 | 250 ± 72 | 0.83 | ||
| αM-/- | |||||||
| PBS | 2 | 2.0 ± 1.2 | 7 ± 3 | 0 ± 0 | NA | ||
| Fuc | 3 | 20.3 ± 2.8 | 177 ± 79 | 97 ± 32 | 1.82 | ||
Genotype and treatment | WBCs, × 106/mL blood | CFUs/mL of blood | |||||
|---|---|---|---|---|---|---|---|
| n of mice | EGFP- | EGFP+ | Ratio | ||||
| Experiment 1 | |||||||
| αM+/+ | |||||||
| PBS | 2 | 3.9 ± 0.1 | 0 ± 0 | 0 ± 0 | NA | ||
| Fuc | 3 | 31.7 ± 6 | 283 ± 18 | 270 ± 32 | 1.04 | ||
| αM-/- | |||||||
| PBS | 2 | 5.0 ± 0.8 | 7 ± 3 | 0 ± 0 | NA | ||
| Fuc | 3 | 25.2 ± 2 | 280 ± 55 | 147 ± 29 | 1.90 | ||
| Experiment 2 | |||||||
| αM+/+ | |||||||
| PBS | 2 | 2.6 ± 0.0 | 0 ± 0 | 0 ± 0 | NA | ||
| Fuc | 3 | 18.2 ± 3.2 | 207 ± 48 | 250 ± 72 | 0.83 | ||
| αM-/- | |||||||
| PBS | 2 | 2.0 ± 1.2 | 7 ± 3 | 0 ± 0 | NA | ||
| Fuc | 3 | 20.3 ± 2.8 | 177 ± 79 | 97 ± 32 | 1.82 | ||
Blood was harvested 2 hours after treatment with fucoidan (100 mg/kg) from EGFP-αM+/+ and EGFP-αM-/- chimeric mice. WBC counts were determined using an automated cell counter. The number of fluorescent and nonfluorescent colonies was determined on day 7 after plating.
Conversion factor: WBC count (1 × 106/mL = 103/μL).
PBS indicates phosphate-buffered saline-treated; NA, not applicable; and Fuc, fucoidan-treated.
Enhanced HPC mobilization in αM–/– mice at suboptimal doses of G-CSF or cyclophosphamide
If αMβ2 played a role in the retention of HPCs in the BM, we would expect an enhanced mobilization in its absence with other mobilizing stimuli. To test this possibility, we induced mobilization with G-CSF, the most common cytokine used to elicit circulating HPCs for clinical transplantation. As shown in Figure 8A, 1 day (2 doses) of G-CSF treatment resulted in few mobilized CFU-Cs and there was no difference in circulating HPCs between αM–/– and αM+/+ mice. However, after 2 days (4 doses) of G-CSF administration, the numbers of circulating HPCs were dramatically increased in αM–/– mice (2.5-fold compared with wild-type counterparts; P = .02). However, following the administration of a maximal stimulus (4 day treatment), the difference between the 2 groups was no longer apparent (Figure 8A).

Increased HPC mobilization in αM–/– mice treated with CY and G-CSF. (A) G-CSF mobilization. αM+/+ (□) and α–/– (▪) mice were treated with G-CSF (250 μg/kg/d) over a period of 1 (n = 8), 2 (n = 8-10), or 4 (n = 5) days. Three hours after the final injection, nucleated cells were isolated from blood and plated to assay CFU-Cs. *P = .02 compared with αM+/+. (B) CY mobilization. αM+/+ (□) and αM–/– (▪) mice were given a single intraperitoneal dose of CY (100 or 200 mg/kg). Nucleated cells from peripheral blood were harvested after 8 days and assayed for CFU-C content. Shown are mean ±SEM numbers of CFU-Cs per milliliter of blood. **P = .01 compared with αM+/+ mice.
Increased HPC mobilization in αM–/– mice treated with CY and G-CSF. (A) G-CSF mobilization. αM+/+ (□) and α–/– (▪) mice were treated with G-CSF (250 μg/kg/d) over a period of 1 (n = 8), 2 (n = 8-10), or 4 (n = 5) days. Three hours after the final injection, nucleated cells were isolated from blood and plated to assay CFU-Cs. *P = .02 compared with αM+/+. (B) CY mobilization. αM+/+ (□) and αM–/– (▪) mice were given a single intraperitoneal dose of CY (100 or 200 mg/kg). Nucleated cells from peripheral blood were harvested after 8 days and assayed for CFU-C content. Shown are mean ±SEM numbers of CFU-Cs per milliliter of blood. **P = .01 compared with αM+/+ mice.
Cyclophosphamide (CY) can also induce a profound HPC mobilization in mice, which peaks from 6 to 8 days after its administration.6 To determine the role of αMβ2 in CY-induced HPC mobilization, we treated αM–/– and αM+/+ animals with either a dose that produces maximal mobilization (200 mg/kg) or a reduced dose (100 mg/kg). In keeping with the above results with G-CSF, there was a 2-fold increase in mobilization when αM–/– mice were treated with a suboptimal dose of CY, whereas no difference was observed when a full dose was administered (Figure 8B). These results are consistent with the notion that HPC mobilization is enhanced when the αMβ2 integrin is absent but that this effect can be overcome by a maximal stimulus. Thus, these data suggest that αMβ2 may serve as a molecular anchor that contributes to the retention of HPCs in the BM.
Discussion
This study clearly shows 2 separate findings: (i) fucoidan binds to αMβ2 and it can release elastase activity from neutrophils, but neither of these activities appear to contribute to its effect on mobilization since fucoidan-induced egress is increased in αM–/– mice and an elastase inhibitor did not influence mobilization; (ii) although we found that plasma CXCL12 levels were slightly increased in αM–/– mice, experiments in αM–/– chimeric mice strongly suggest that the enhanced mobilization in αM–/– mice is intrinsic to hematopoietic cells. The fact that a similar increase in mobilization was observed with other stimuli (CY and G-CSF) is consistent with the idea that αMβ2 may act as an adhesive anchor that contributes to the retention of HPCs in the BM during enforced egress.
Although αMβ2 was initially discovered as a macrophage complement receptor,10 it ranks among the most pleiotropic cell surface adhesion molecules that can mediate interactions with platelets,39 coagulation proteins,40,41 complement components,10 proteolytic enzymes,11 carbohydrates,15 and cellular receptors.14 The interactions described here between αMβ2 expressed on myeloid cells and soluble or immobilized fucoidan are consistent with the recent work of Zen et al17 who showed a direct binding between fucoidan and purified immobilized αMβ2.
It is interesting that αM contains a carbohydrate binding (lectin) domain located outside the “inserted” I domain.42 Like all integrins, a change in the conformation of extracellular domains is critical for αMβ2-mediated adhesion. In particular, the I domain with its metal ion-dependent adhesion site (MIDAS) has been shown to be crucial for interaction with counterreceptors.42 In contrast to the previously described interaction with heparin,13 we show here that myeloid cells (and αMβ2) do not require prior activation to bind fucoidan (Figure 3). This suggested 2 possibilities: (1) fucoidan may independently activate myeloid cells prior to binding αMβ2; or (2) fucoidan may bind an activation-independent epitope on αMβ2. While our data suggest that the former is less likely, the latter would be consistent with previous work. Binding of soluble β-glucan or yeast cell walls to the αM lectin domain was shown to generate high-affinity binding conformation of the I domain.43,44 Consistent with its ability to strictly bind activated αMβ2, the heparin binding site likely resides in the I domain.13 In the present study, the inhibition by Mab M1/70 of BMNC adhesion to immobilized fucoidan suggests that fucoidan also interacts with the I domain. Thus, it is conceivable that fucoidan might first bind αM outside the I domain (possibly in the lectin domain) and that this binding might then induce allosteric conformational changes leading to high-affinity MIDAS-mediated integrin binding of the same or other fucoidan molecules to the I domain. Although the inhibition of αMβ2 by fucoidan raises the possibility that this interaction may contribute to the powerful effects of the sulfated polysaccharide on leukocyte adhesion in vivo,21,45 we found no evidence that it was involved in the mobilization of HPCs from the BM.
Since fucoidan could bind αMβ2 without exogenous activation, we evaluated whether it could activate myeloid cells and induce the release of serine proteases. Recent studies indeed indicate that neutrophil serine proteases may play prominent roles in HPC mobilization.5,6,30,36 Although fucoidan induced a profound release of both elastase and cathepsin G in the supernatant, we did not observe neutrophil degranulation (as measured by the lack of up-regulation of αMβ2 expression and L-selectin shedding from the cell surface) or activation (as measured by induction of oxidative burst). Since elastase was previously shown to be presented by αMβ2 on the cell surface,11 we evaluated the ability of αM–/– cells to release elastase and found that it was not influenced by αMβ2 expression. Although the exact origin (granular vs membrane associated) of released neutrophil proteases following fucoidan treatment is currently unclear, we present evidence that elastase activity is dispensable since an elastase inhibitor previously shown to efficiently inhibit mobilization by G-CSF30 did not impact HPC egress triggered by fucoidan.
Because accumulating data suggest that CXCL12 plays a key role in HPC trafficking,46,47 we assessed CXCL12 levels in the plasma and BM of αM–/– and αM+/+ mice in fucoidan-treated or after control PBS administration. In agreement with a previous study,27 we found that fucoidan significantly increased plasma CXCL12 levels in wild-type mice and that CXCL12 plasma elevation was significantly more pronounced in αM–/– mice (Figure 5). In addition, we found that soluble fucoidan (a negatively charged polysaccharide that can interact with basic residues of CXCL1248 ) appears to enhance CXCL12-mediated migration likely by presentation of multiple CXCL12 molecules to HPCs. We believe that this multivalent presentation of CXCL12 by fucoidan in the plasma is likely to play a role in the attraction of HPCs in the blood. Since αMβ2 is a scavenger receptor able to remove “foreign” glycosylated structures from the circulation,49 it is conceivable that it participates in the clearance of fucoidan-CXCL12 complexes, thus explaining the higher plasma levels of CXCL12 in αM–/– mice.
However, our data suggest that the increased mobilization in αM–/– is intrinsic to hematopoietic cells and does not result from changes in the microenvironment. Indeed, using a competitive approach in which both αM+/+ and αM–/– cohabited in the bone marrow (therefore subjected to the same microenvironment and chemoattractants), we found that αM-deficient progenitors were preferentially mobilized over those αM-sufficient. These results strongly suggest that αMβ2 plays a direct role in HPC retention in the bone marrow. Consistent with this notion, the phenotype was not restricted to a single mode of mobilization, and significant increases in HPC mobilization in αM–/– mice were found using other methods. It is interesting to note, however, that the αMβ2-mediated anchorage was no longer detectable when stronger mobilizers (full doses of CY or G-CSF) were used, suggesting that powerful stimuli could overcome αMβ2-mediated retention. In agreement with this observation, Velders et al7 reported enhanced HPC mobilization in mice treated with an anti-αM antibody with low doses of G-CSF. We have previously studied the role of L-selectin in this activity and showed that fucoidan-mediated egress proceeded normally in L–/– mice, but using the same strategy described here (Figure 7) to generate chimeric mice harboring both L+/+ and L–/– cells, we found that L+/+ cells were predominantly mobilized by fucoidan.25 Of note, we have recently generated mice lacking both L-selectin and αM to investigate their combined roles and found that fucoidan-mediated HPC mobilization in doubly-deficient mice was increased to levels similar to those of αM–/– animals (A.H., A.J.P., and P.S.F., unpublished data, February 2003).
It is also important to emphasize that the studies described here involve the migration of colony-forming colonies which assay progenitors that express higher levels of αMβ2 than do hematopoietic stem cells (HSCs). Long-term self-renewing hematopoietic stem cells (LT-HSCs) in the bone marrow were reported to express little or no αMβ2,50 but its expression can be induced in situations in which HSCs undergo active cell cycling such as in the fetal liver8 and after 5-fluorouracil treatment.9 Following CY and G-CSF stimulation, the stem cell activity in the bone marrow and spleen is found in both αMβ2-positive and -negative subsets, but subsets expressing αMβ2 were enriched in non–self-renewing multipotent progenitors.51 Given the higher expression of αMβ2 among “activated” HSCs,9 it is possible that αMβ2 might participate in the migration and/or redistribution of HPCs/HSCs among hematopoietic niches in the bone marrow.
Prepublished online as Blood First Edition Paper, April 20, 2004; DOI 10.1182/blood-2003-10-3702.
Supported by the National Institutes of Health grant DK-56638 (P.S.F.) and a grant from the Cooley's Anemia Foundation (A.H.).
A.H. and A.J.P. contributed equally to this study.
Presented in part at the 42nd annual meeting of the American Society of Hematology, San Francisco, CA, December 2000.52
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.
We thank Drs Richard Hynes, Tanya Mayadas, and Sergio Lira for providing transgenic animals. We also thank Dr Isabelle Petit for her helpful advice with the CXCL12 ELISA assay.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal