

TO THE EDITOR:
VEXAS syndrome (vacuoles in myeloid progenitors, E1 ubiquitin-activating enzyme, X-linked, autoinflammatory manifestations, and somatic) is the consequence of the expansion of hematopoietic stem and/or progenitor cells with somatically acquired UBA1 (ubiquitin-like modifier activating enzyme 1) mutations.1,2 Patients present with a variety of autoinflammatory manifestations, and approximately half of them have an associated hematological malignancy, mainly myelodysplastic syndrome (MDS) and/or monoclonal gammopathy.3,4 Long-term use of high doses in this steroid-dependent disease is often associated with unacceptable side effects. Retrospective studies have underlined the poor response of VEXAS patients to a variety of therapeutic strategies, except for a few patients exposed to Janus kinase inhibitors (JAKi).2,5 Here, we present the results of a multicenter international retrospective analysis of VEXAS patients treated with different JAKi to better characterize safety and efficacy profiles.
Thirty patients with genetically proven VEXAS syndrome treated with JAKi (ruxolitinib [n = 12], tofacitinib [n = 11], baricitinib [n = 4], or upadacitinib [n = 3]) between April 2018 and February 2022 were included in 10 hospitals from France, the United States, and Portugal. Patients empirically treated with JAKi before a formal retrospective diagnosis of VEXAS were identified after a call for cases among participants of the French VEXAS Group and clinicians from Mayo Clinic and the National Institutes of Health. We also included patients treated by JAKi after the publication of our first series describing therapeutic outcomes in VEXAS patients.2 Fourteen patients had concomitant myeloid neoplasia (MN), mostly MDS (supplemental Table 1). Clinical manifestations of VEXAS involved the skin (n = 26), joints (n = 25), and lungs (n = 17), with persistent fever in 24 patients and venous thromboembolism (VTE) in 10 patients (supplemental Table 1). High throughput sequencing of myeloid malignancy driver genes was available in 17 patients, including 10 MN patients (supplemental Table 2). Only 1 non-MN patient had additional mutations (in DNMT3A and KDM6A), whereas half of the MN patients had additional mutations (involving DNMT3A [n = 3], TET2 [n = 1], JAK2 [n = 1], and CALR [n = 1]). Patients with MN had a lower incidence of fever at diagnosis (57% vs 94%; P = .041), of prior history of thrombotic events (21% vs 50%; P = .034), and a trend for higher frequency of red blood cell (RBC) transfusion dependency (50% vs 17%; P = .074) (supplemental Table 3). Patients had received a median of 2.5 immunosuppressive or immunomodulatory treatments (range, 0-9) before the start of a JAKi (supplemental Table 4).
An overview of patient follow-up is provided in supplemental Figure 1. Overall, 15 out of 30 (50%) patients had a clinical response (CR) after 1 month of treatment. Biological response (BR), defined as a >50% reduction of C reactive protein level, was observed in 20 patients, including 11 complete biological response (CBR; normalization of C reactive protein level) (Figure 1; supplemental Figure 1). After 3 months of treatment, CR and BR rates were 57.1% (12/21) and 53.6%, respectively. After 6 months, 11 patients remained on treatment, of which 9 and 3 were in CR and CBR, respectively. The median duration of CR was not reached. A subgroup analysis showed higher response rates in patients treated with ruxolitinib (n = 12) than in those treated with other JAKi (n = 18), at 1 (CR, 67% vs 38%; P = .13), 3 (CR, 83 vs 18%; P = .001), and 6 months (CR, 87% vs 11%; P = .002), respectively (Figure 1). JAKi treatment was discontinued in 9 patients receiving JAKi other than ruxolitinib because of lack of efficacy (4 before 1 month, and 5 before 3 months).

Antiinflammatory effects of JAKi in patients with VEXAS syndrome. (A) Overall clinical response rates of patients treated with JAKi during 1, 3, or 6 months; results are presented for the whole cohort (left), for patients receiving other JAKi than ruxolitinib (middle), or for patients receiving ruxolitinib (right). (B) Overall biological response rates of patients treated with JAKi during 1, 3, or 6 months; results are presented for the whole cohort (left), for patients receiving other JAKi than ruxolitinib (middle), or for patients receiving ruxolitinib (right).
Antiinflammatory effects of JAKi in patients with VEXAS syndrome. (A) Overall clinical response rates of patients treated with JAKi during 1, 3, or 6 months; results are presented for the whole cohort (left), for patients receiving other JAKi than ruxolitinib (middle), or for patients receiving ruxolitinib (right). (B) Overall biological response rates of patients treated with JAKi during 1, 3, or 6 months; results are presented for the whole cohort (left), for patients receiving other JAKi than ruxolitinib (middle), or for patients receiving ruxolitinib (right).
Ruxolitinib efficacy was similar in patients with or without associated MN (supplemental Figure 2). With a median follow-up of 6.9 months (range, 1-41), 9 out of 12 (75%) patients treated with ruxolitinib were still receiving the drug vs only 5 out of 18 (28%) patients treated with another JAKi (supplemental Figure 1). Ruxolitinib dosage was increased during the treatment period, from a mean starting dose of 15 mg per day (range, 10-20 mg per day) to a mean dose at the last follow-up of 25.4 mg per day (range, 10-50 mg per day). There was no dose modification for other JAKi during the treatment period.
The median time to the next line of treatment was not reached in patients on ruxolitinib but was 3.3 months (95% confidence interval, 1.3-12.1; P < .001) in patients treated with other JAKi (Figure 2A). Among patients still on treatment at 6 months, the median steroid dose reduction was 83.6% and 75% for patients treated with ruxolitinib and other JAKi, respectively (Figure 2B). At last follow-up, 3 patients were off steroids (ruxolitinib [n = 2] and upadacitinib [n = 1]). Prognostic factors of response to JAKi are shown in supplemental Figure 3.
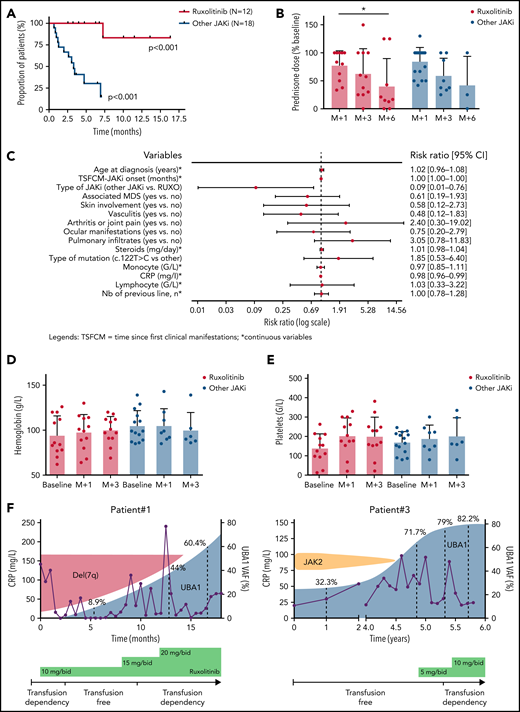
Clinical (and hematological) outcome of patients with VEXAS syndrome treated with JAKi. (A) Kaplan-Meier representation of the time to next treatment in patients treated by ruxolitinib or other JAKi. (B) Prednisone dose (percentage of baseline) after 1, 3, and 6 months in patients treated with ruxolitinib or other JAKi. (C-D) Hemoglobin and platelet count evolution at baseline and after 1 and 3 months of ruxolitinib in patients treated with ruxolitinib or other JAKi. (E) UBA1 fish plot of 2 patients treated with ruxolitinib.
Clinical (and hematological) outcome of patients with VEXAS syndrome treated with JAKi. (A) Kaplan-Meier representation of the time to next treatment in patients treated by ruxolitinib or other JAKi. (B) Prednisone dose (percentage of baseline) after 1, 3, and 6 months in patients treated with ruxolitinib or other JAKi. (C-D) Hemoglobin and platelet count evolution at baseline and after 1 and 3 months of ruxolitinib in patients treated with ruxolitinib or other JAKi. (E) UBA1 fish plot of 2 patients treated with ruxolitinib.
A significant increase in hemoglobin levels was observed in patients treated with ruxolitinib (mean hemoglobin variation at 3 months, +0.9 g/L) compared with those treated with other JAKi (P = .031) (Figure 2C). Of the 14 patients with MN, 7 were RBC transfusion-dependent at JAKi onset, and 4 (all treated with ruxolitinib) achieved RBC transfusion independence after 1 month (supplemental Figure 4). Similarly, a significant increase in platelet counts was seen in patients receiving ruxolitinib (P = .028) (Figure 2D). Worsening of cytopenias, especially anemia, was not uncommon during the first weeks of treatment. Supportive care recommendations for VEXAS patients treated with ruxolitinib are provided in the supplemental material. MDS progression was observed in 2 patients treated with ruxolitinib.
Longitudinal monitoring of the clonal burden of UBA1-mutated cells was performed by measuring the variant allele frequency of UBA1 mutation in peripheral blood of 2 patients treated with ruxolitinib. An expansion of the UBA1-mutated clone was observed in both, which outcompeted a del(7q) or a JAK2 V617F clone in each patient (Figure 2E).
The most frequent adverse events were infections (36.7%) and thromboembolic complications (20%) (detailed in the supplemental Material). Overall, 3 (10%) patients died: 1 from legionellosis (treated with tofacitinib), 1 from colon cancer progression (treated with ruxolitinib), and 1 from an undetermined cause (treated with ruxolitinib).
This retrospective multicenter study shows that JAKi, especially ruxolitinib, are effective therapeutic options in patients with VEXAS syndrome. The better efficacy of ruxolitinib over other JAKi could potentially be due to its target specificity, as ruxolitinib and baricitinib inhibit mainly JAK1 and JAK2, while tofacitinib and upadacitinib preferentially target JAK1 and JAK3.6 Moreover, ruxolitinib and baricitinib also have enhanced inhibitory activity on TYK2, a member of the JAK family of receptor-associated tyrosine kinases ubiquitously expressed in blood cells.7,8 The better efficacy of ruxolitinib might further be explained by its broader dosing range, enabling progressive dose increase, which is not possible with other JAKi. Alternatively, ruxolitinib could be preferentially effective in VEXAS patients with underlying MN. Given the retrospective nature of this study, we cannot draw definitive conclusions regarding the different hypotheses explaining the superiority of ruxolitinib, but our results suggest that ruxolitinib could be the preferred JAKi for VEXAS syndrome patients.
As the main adverse events reported in this series are known complications of VEXAS, the causal role of JAKi in their occurrence is unclear. Infections have been described in most clinical trials of JAKi, mostly due to the reactivation of herpes family viruses.9,10 Severe bacterial and nonherpes viral infections were the most prominent events, suggesting that the immunodepression of VEXAS patients is not reversed by JAKi treatment. The other major complication was thrombosis, which was highly prevalent in VEXAS patients in previous cohorts4 and this series, even before JAKi initiation. Whether JAKi had a causal role in these VTEs is an open question. Recently, JAKi and, more specifically, tofacitinib and baricitinib have been incriminated for potential higher risk of VTE,11 but a large meta-analysis of 42 studies did not support evidence for higher risk of VTE in JAKi-treated patients.12 Altogether, even if JAKi provides rapid and sustained clinical response, VEXAS patients still continue to experience recurrent infectious and thrombotic events.
Finally, the observation of UBA1 clonal expansion in both patients with available data strongly suggests that ruxolitinib has mainly a suspensive effect and cannot cure VEXAS patients. While waiting for prospective clinical trials, these data collected internationally from therapeutic interventions in VEXAS patients offer some of the only available information to guide therapeutic decisions in this potentially devastating disease.
Acknowledgment
This study was not funded by a specific grant.
Authorship
Contribution: M.H. designed the project, acquired and analyzed the data, and wrote the paper; M.A.F., M.T.K., T.B., M.G.-V., A.M., H.C., G.F., F.B., L.G., B.B., P.H., G.V., A.B.B., J.G., G.L.G., A.B., K.J.W., and T.A.K. acquired data and treated patients; P.C.G., B.A.P., D.B.B., Y.J., and P.F. acquired data and reviewed manuscript; and P.S. acquired and analyzed the data, reviewed the manuscript, and gave final approval.
Conflict-of-interest disclosure: P.H. consults for Novartis. K.J.W. receives clinical trial support from Eli Lilly. The remaining authors declare no competing financial interests.
Correspondence: Pierre Sujobert, Service d'Hématologie Biologique, Centre Hospitalier Lyon Sud., 165 chemin du grand Revoyet, 69310 Pierre Bénite, France; e-mail: pierre.sujobert@chu-lyon.fr.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.

