Key Points
CRISPR-Cas9 screening identified RIOK2 as a potential novel dependency target in AML.
Loss of RIOK2 catalytic activity led to stalled protein synthesis, ribosome degradation, and cell death.

Abstract
Novel therapies for the treatment of acute myeloid leukemia (AML) are urgently needed, because current treatments do not cure most patients with AML. We report a domain-focused, kinome-wide CRISPR-Cas9 screening that identified protein kinase targets for the treatment of AML, which led to the identification of Rio-kinase 2 (RIOK2) as a potential novel target. Loss of RIOK2 led to a decrease in protein synthesis and to ribosomal instability followed by apoptosis in leukemic cells, but not in fibroblasts. Moreover, the ATPase function of RIOK2 was necessary for cell survival. When a small-molecule inhibitor was used, pharmacological inhibition of RIOK2 similarly led to loss of protein synthesis and apoptosis and affected leukemic cell growth in vivo. Our results provide proof of concept for targeting RIOK2 as a potential treatment of patients with AML.
Introduction
Despite significant advances in understanding the underlying molecular pathology and genetic landscape of acute myeloid leukemia (AML), as well as the recent approval of several drugs for the clinical treatment of AML, improving the survival outcome for patients is an ongoing challenge.
Ribosome biogenesis is a complex, multistep process that involves >200 nonribosomal proteins and has been investigated as a potential target for cancer treatment.1,2 Because rapidly dividing cancer cells have an increased demand for protein synthesis, hyperactivation of ribosome biogenesis is a commonly occurring feature of cancer cells.3 Rio-kinase 2 (RIOK2) is an ATPase that plays a role in the nuclear export and final cytoplasmic maturation of the 40S ribosomal subunit in yeast and human cells.4,5 In concert with other 40S assembly factors, RIOK2 is also involved in preventing premature translation initiation during 40S ribosomal subunit maturation by obstructing binding sites for the translation initiation factors eIF1 and eIF1A.6 High expression of RIOK2 has been linked to decreased overall survival in non–small cell lung cancer.7 Moreover, knockdown of Riok2 leads to dramatic growth reduction of Drosophila neoplastic glial cells, but not of normal glia.8 Although there is a basic understanding that RIOK2 is involved in regulating ribosome biogenesis and translation, the molecular mechanisms by which it can contribute to cancer remain largely unexplored.
Several approaches targeting protein synthesis as a means of treating cancer have been suggested in recent years.9,10 One common approach involves targeting the PI3K/AKT/mTOR pathway, which is one of the central pathways involved in the regulation of protein synthesis and translation initiation, is constitutively active in 50% to 80% of all AML cases, and is often associated with a poor clinical outcome.11 However, phase 1 and 2 trials targeting mTOR signaling have failed to show meaningful effectiveness in the treatment of patients with AML.12 The identification of novel approaches that target protein synthesis is therefore of high importance.
We performed a kinome-wide CRISPR screening in murine MLL-AF9 translocated leukemic cells to identify novel kinase targets for the treatment of AML. We identified RIOK2 as a potential novel target and found that it was required for the growth of leukemic cells both in vitro and in vivo. By the use of a recently described small-molecule inhibitor of RIOK2,13 we provide proof of concept for targeting RIOK2 in AML.
Materials and methods
Kinome-wide CRISPR-Cas9 screening
A list of 545 murine kinases compiled from the literature was used to generate a kinome-wide domain-focused CRISPR/Cas9 library. The mm10 PFAM domain annotations were used to map the kinase domain, and 5 to 10 single guide RNAs (sgRNAs) targeting within this domain were selected based on published design strategies.14 An oligo pool consisting of 6237 60-bp oligos targeting all 545 kinases, as well as 10 positive and 1000 negative controls, was synthesized by CustomArray. The library was amplified by polymerase chain reaction (PCR), cloned into the U6-sgRNA-SFFV-puro-P2A-EGFP plasmid, and sequence verified by next-generation sequencing (NGS), as described.15 A lentivirus pool was generated from the plasmid library and used to transduce duplicate cultures of MA9 cells with a transduction efficiency of 30% and an initial coverage of 1000×. After 48 hours of puromycin selection (2 µg/mL) an initial sample was taken, and the cultures were grown for 10 additional doublings. Genomic DNA was isolated from cells harvested at both time points. Integrated sgRNAs were PCR amplified and sequenced by NGS. Sequencing results were analyzed using DEseq2,16 and gRNAs were depleted or enriched at the end point were identified as described.15
EdU labeling–based cell cycle analysis
Cells transduced with lentiviruses expressing sgRNAs targeting Riok2, Rps19, or a nontargeting control were selected by the addition of puromycin (2 µg/mL) for 72 hours, 2 days after transduction, or treated with RIOK2i for 72 hours. EdU labeling was performed with the Click-iT EdU Alexa Fluor 488 Flow Cytometry Assay Kit (C10425; Thermo Fisher Scientific), according to the manufacturer’s instructions, with 1 µM EdU and a labeling time of 45 minutes. The cells were stained with 4′,6-diamidino-2-phenylindole (1 µg/mL) before analysis. Flow cytometry was performed on BD FACS AriaIII (BD Biosciences), and data analysis was performed with FlowJo software.
In vitro OP-puro labeling
Cells were transduced with lentiviruses expressing sgRNAs against Riok2, Rps19, or NC-1 cloned into the pLKO5-sgRNA-EFS-tRFP657 vector. RFP+ cells were sorted on a FACS AriaIII, 2 days after transduction. O-propargyl-puromycin (OP-puro) labeling was performed 48 hours after sorting with the Protein Synthesis Assay Kit (ab235634; Abcam), according to the manufacturer’s instructions. The experiment was performed in triplicate, starting from 3 independently transduced wells. Cells were labeled with OP-puro for 45 minutes before fixation. With the same protocol, OP-puro labeling was also performed on RIOK2i-treated cells 48 hours after treatment, using either 300 nM or 1 µM of RIOK2i.
Animal studies
All mouse experiments were approved by the Danish Animal Ethics Committee (license 2017-15-0201-01176). Detailed information on animal housing and additional experimental procedures can be found in the supplemental Information (available on the Blood Web site).
RNA-sequencing
Riok2fl/fl R-CreER MA9 cells were seeded in triplicate with either 250 000 cells per well (ethanol [EtOH] treated) or 400 000 cells per well (OHT treated) in 3 mL medium. The cells were collected 72 hours after addition of EtOH or OHT at a final concentration of 1 µM. MA9 wild-type (WT) cells were treated with RIOK2i for 72 hours in triplicate experiments, at a concentration of 1 µM, or with dimethyl sulfoxide (DMSO) as the control. RNA-extraction was performed with the RNeasy Mini Kit (74106; Qiagen). Before library preparation, RNA-Integrity was assessed on the Bioanalyzer 2100 (Agilent) on RNA HS chips (5067-1513; Agilent). Only nondegraded RNA (RIN >7) was used for subsequent library preparation. RNA libraries were made by using the TruSeq RNA Library Prep Kit (RS-122-2001; Illumina). The size and quality of the library were assessed on a DNA HS chip (5067-4627; Agilent). Pooled libraries were sequenced on the Illumina NextSeq 500 system. Computational pipelines for the analysis of the RNA-sequencing (RNA-Seq) data can be found in the supplemental Information.
Results
RIOK2 is essential for the survival of AML cells in vitro
We performed a domain-focused, kinome-wide CRISPR screening of mouse MLL-AF9 (MA9) cells as a model system (Figure 1A; supplemental Figure 1A) to identify novel kinase targets in AML. Genes coding for several kinases that were previously identified as being important for AML maintenance such, as Atr,17Ttk,18 and Cdk1,19 were among the most depleted (supplemental Data File 1). We also identified guide RNAs targeting the Riok2 kinase gene as among the highest depleted (Figure 1B; supplemental Table 1). sgRNAs targeting RIOK2 were also reported to be depleted in a screening for the proliferation of human AML cell lines (supplemental Figure 1C).20 Interestingly, by analyzing patient samples from the Beat AML cohort,21 we found that RIOK2 showed significantly higher expression in different AML subtypes compared with healthy bone marrow mononuclear cells (supplemental Figure 1B).
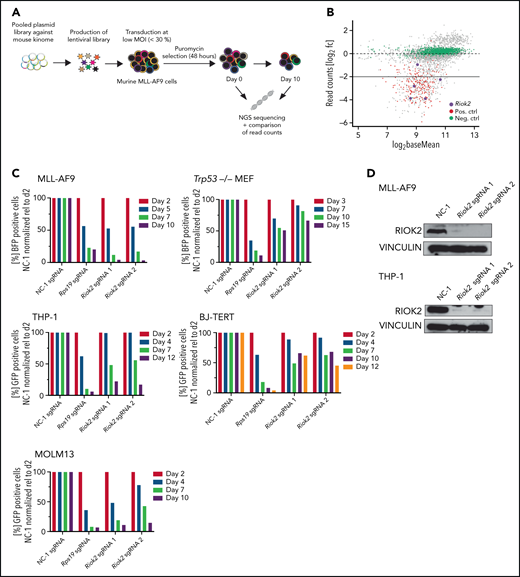
Identification and in vitro validation of RIOK2 dependency in AML. (A) Experimental overview of the kinome-wide, domain-focused CRISPR screening. Murine cells harboring an MLL-AF9 translocation were transduced with a lentiviral sgRNA library followed by puromycin selection. NGS was performed on samples immediately after and 10 days after the selection. (B) DeSeq2 analysis of the screening results. sgRNAs for positive control genes are depicted in red, negative control sgRNAs in green, and sgRNAs targeting Riok2 in purple. (C) Competition-based proliferation assays in murine MLL-AF9 and Trp53−/− MEFs and human THP-1, BJ-TERT and MOLM13 cell lines using an sgRNA plasmid containing a BFP or GFP fluorophore. The percentage of positive cells was quantified by FACS and normalized to the nontargeting control, relative to the initial time point for each of the indicated sgRNAs. Data from a representative experiment are shown. (D) Western blots for murine and human RIOK2 4 days after sgRNA transduction. Cells were selected by puromycin for 3 days, beginning 24 hours after transduction with the indicated sgRNAs. NC-1, nontargeting control 1.
Identification and in vitro validation of RIOK2 dependency in AML. (A) Experimental overview of the kinome-wide, domain-focused CRISPR screening. Murine cells harboring an MLL-AF9 translocation were transduced with a lentiviral sgRNA library followed by puromycin selection. NGS was performed on samples immediately after and 10 days after the selection. (B) DeSeq2 analysis of the screening results. sgRNAs for positive control genes are depicted in red, negative control sgRNAs in green, and sgRNAs targeting Riok2 in purple. (C) Competition-based proliferation assays in murine MLL-AF9 and Trp53−/− MEFs and human THP-1, BJ-TERT and MOLM13 cell lines using an sgRNA plasmid containing a BFP or GFP fluorophore. The percentage of positive cells was quantified by FACS and normalized to the nontargeting control, relative to the initial time point for each of the indicated sgRNAs. Data from a representative experiment are shown. (D) Western blots for murine and human RIOK2 4 days after sgRNA transduction. Cells were selected by puromycin for 3 days, beginning 24 hours after transduction with the indicated sgRNAs. NC-1, nontargeting control 1.
To further test the requirement of RIOK2 for leukemic cell proliferation, we performed proliferation-based competition assays with sgRNAs targeting Riok2 in MA9 cells, and as a control for specificity, in immortalized Trp53−/− mouse embryonic fibroblasts (MEFs; Figure 1C). Targeting Riok2 led to a strong depletion of cells that was similar to the depletion of the common essential gene Rps19 in MLL-AF9 cells, but the effect was less pronounced in the Trp53−/− MEFs (Figure 1C-D). We observed a similar differential effect in human AML cell lines and in a human fibroblast cell line (Figure 1C-D). Interestingly, RIOK1, a close homologue of RIOK2, did not show a differential requirement in leukemia cells and in fibroblasts, being essential in both types of cells (supplemental Figure 1D). The stabilization of p53 in response to the inhibition of MDM2 by free ribosomal proteins has been shown to be an important mechanism through which ribosomal stress induces apoptosis.22,23 To test whether the effect of RIOK2 loss is dependent on p53, we generated Trp53−/− MA9 cells (supplemental Figure 1E). The p53 knockout MA9 cells were as sensitive to loss of RIOK2 as p53 WT cells were (supplemental Figure 1F), demonstrating that RIOK2 is necessary, independent of p53, and also suggesting that the milder proliferation defects observed in the Trp53−/− MEFs were not related to loss of p53. Taken together, our results show that RIOK2 is essential for the proliferation of AML cells in vitro.
RIOK2 is essential for AML maintenance in vivo
To further investigate the role of RIOK2 in AML, we generated an MLL-AF9 cell line with the conditional inactivation of Riok2 (Riok2fl/flROSA26::CreER [R-Cre]) by transforming c-Kit+ bone marrow cells derived from a Riok2fl/fl R-Cre mouse (Figure 2A; supplemental Figure 2A). Activation of the Cre recombinase by addition of 4-hydroxytamoxifen (OHT) to the culture medium led to efficient recombination of the Riok2 locus and loss of expression of the RIOK2 protein (Figure 2B-C). Similar to the CRISPR-Cas9–mediated deletion of Riok2, Cre-mediated loss of Riok2 led to a strong decrease in cell proliferation (Figure 2D). To test whether RIOK2 is also necessary for maintenance of leukemic cells in vivo, we transplanted the Riok2fl/fl R-Cre MA9 cells into sublethally irradiated mice and injected the transplant recipients with tamoxifen or oil, beginning 14 days after transplantation and continuing for the next 5 days (Figure 2E). The mice injected with oil died within 28 to 56 days, whereas none of the mice injected with tamoxifen died within the time frame of the experiment (70 days; Figure 2F). Spleen weight analysis after termination of the experiment showed no significant increase in spleen weight in the tamoxifen-treated mice, whereas spleens of the oil-treated mice were significantly enlarged (Figure 2G). Taken together, these results demonstrate that RIOK2 is essential for AML maintenance in vivo.
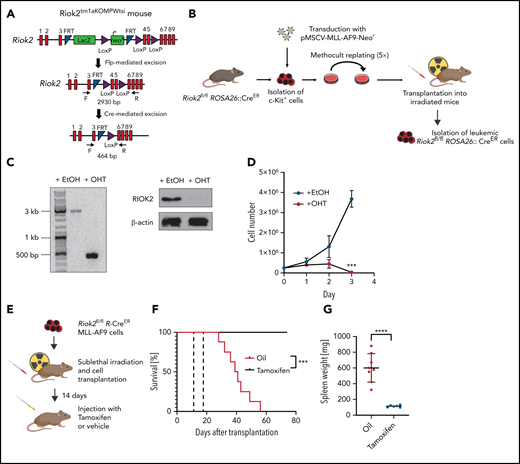
RIOK2 is required for maintaining AML in vivo. (A) Genetic locus and crossing strategy of the Riok2tm1aKOMPWtsi mouse. After rederivation, the mice were crossed with FlpE mice to excise the LacZ+neoR cassette. The progeny of this cross was then crossed with ROSA26::CreERT mice to induce Cre-mediated excision of exons 4 and 5 of Riok2 after tamoxifen treatment. (B) Experimental outline of the Riok2fl/fl R-CreER MLL-AF9 cell line generation. c-Kit+ cells were isolated from a C57/BL6 carrying homozygous floxed Riok2 alleles and transduced with a retrovirus expressing the MLL-AF9 oncogene. After 5 rounds of replating, the cells were transplanted into sublethally irradiated mice and isolated after the mice developed full-blown leukemia. (C) Agarose gel showing PCR products from the amplified region of the floxed Riok2 locus after a 72-hour treatment of Riok2fl/fl R-CreER MLL-AF9 cells with EtOH or OHT (left). Western blot showing RIOK2 protein levels in Riok2fl/fl R-CreER MLL-AF9 cells 72 hours after addition of EtOH or OHT to the culture medium (right). (D) Growth curve of MA9 Riok2fl/fl R-CreER cells after OHT or EtOH addition to the culture medium (n = 3 biological replicates). Multiple unpaired Student t tests were performed to assess statistical significance between the OHT- and EtOH-treated cells. ***P < .001. Error bars represent standard deviation. (E) Experimental overview testing the role of Riok2 in vivo. Riok2fl/fl R-CreER MLL-AF9 cells were transplanted into sublethally irradiated B6/SJL mice. Fourteen days after transplantation, mice were injected with either tamoxifen or oil and monitored for survival. (F) Survival curve of 14 B6-SJL mice receiving transplants of with MA9 Riok2fl/fl R-CreER cells. Dotted lines indicate the time (days 14-19) when the mice were injected daily with either tamoxifen (n = 6) or oil (n = 8). A log-rank (Mantel-Cox) test was performed to assess differences in survival (P = .0003). (G) Spleen weights of mice at the time of death or experimental end point (day 74). Error bars represent standard deviation (SD). A Student t test was performed to assess significance. ****P < .0001.
RIOK2 is required for maintaining AML in vivo. (A) Genetic locus and crossing strategy of the Riok2tm1aKOMPWtsi mouse. After rederivation, the mice were crossed with FlpE mice to excise the LacZ+neoR cassette. The progeny of this cross was then crossed with ROSA26::CreERT mice to induce Cre-mediated excision of exons 4 and 5 of Riok2 after tamoxifen treatment. (B) Experimental outline of the Riok2fl/fl R-CreER MLL-AF9 cell line generation. c-Kit+ cells were isolated from a C57/BL6 carrying homozygous floxed Riok2 alleles and transduced with a retrovirus expressing the MLL-AF9 oncogene. After 5 rounds of replating, the cells were transplanted into sublethally irradiated mice and isolated after the mice developed full-blown leukemia. (C) Agarose gel showing PCR products from the amplified region of the floxed Riok2 locus after a 72-hour treatment of Riok2fl/fl R-CreER MLL-AF9 cells with EtOH or OHT (left). Western blot showing RIOK2 protein levels in Riok2fl/fl R-CreER MLL-AF9 cells 72 hours after addition of EtOH or OHT to the culture medium (right). (D) Growth curve of MA9 Riok2fl/fl R-CreER cells after OHT or EtOH addition to the culture medium (n = 3 biological replicates). Multiple unpaired Student t tests were performed to assess statistical significance between the OHT- and EtOH-treated cells. ***P < .001. Error bars represent standard deviation. (E) Experimental overview testing the role of Riok2 in vivo. Riok2fl/fl R-CreER MLL-AF9 cells were transplanted into sublethally irradiated B6/SJL mice. Fourteen days after transplantation, mice were injected with either tamoxifen or oil and monitored for survival. (F) Survival curve of 14 B6-SJL mice receiving transplants of with MA9 Riok2fl/fl R-CreER cells. Dotted lines indicate the time (days 14-19) when the mice were injected daily with either tamoxifen (n = 6) or oil (n = 8). A log-rank (Mantel-Cox) test was performed to assess differences in survival (P = .0003). (G) Spleen weights of mice at the time of death or experimental end point (day 74). Error bars represent standard deviation (SD). A Student t test was performed to assess significance. ****P < .0001.
The ATPase function of RIOK2 is essential for AML cell survival
In addition to its kinase domain, RIOK2 contains an N-terminal winged helix-turn-helix (wHTH) domain that is thought to have a role in rRNA binding and a C-terminal tail region containing numerous phosphorylation sites.24,25 To determine which parts of RIOK2 are functionally required, we generated MA9 cell lines expressing a control vector harboring a stuffer luciferase protein, WT human RIOK2, or different mutations, including a cell line carrying K123A and D246A, which have been described to be essential for adenosine triphosphate (ATP) hydrolysis4; a second line lacking the N-terminal wHTH domain; and a third one lacking the C-terminal tail region (Figure 3A; supplemental Figure 2A). Although expression of WT RIOK2 completely rescued the proliferation defects (Figure 3B) caused by deleting endogenous Riok2, the 3 mutant lines all failed to do so, demonstrating the functional requirement of ATPase activity and the wHTH and C-terminal regions for survival of AML. These results were further validated in proliferation assays, in which ectopic expression of WT RIOK2, but not the RIOK2K123A, D246A mutant, rescued the growth defects induced by deletion of endogenous Riok2 (Figure 3C; supplemental Figure 2B).
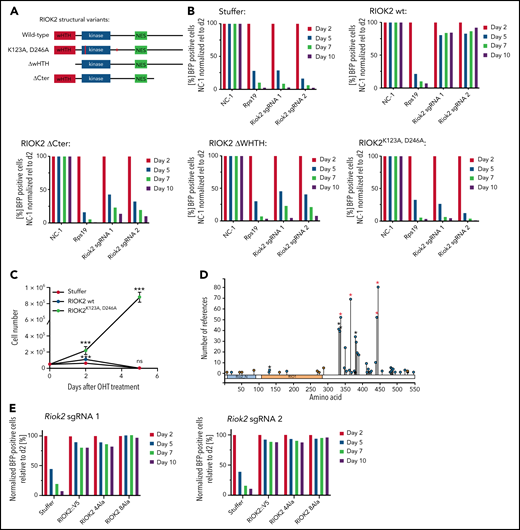
The ATPase function of RIOK2 is essential for supporting leukemia proliferation. (A) Overview of the different RIOK2 mutants used for structure-function analysis. NES, nuclear export signal; wHTH, winged helix-turn-helix domain. (B) Competition-based proliferation assays in murine MLL-AF9 cells expressing the depicted RIOK2 mutants. Expression of luciferase (stuffer) was used as the negative control. MA9 cells were transduced with an sgRNA plasmid containing a BFP fluorophore to knock out endogenous Riok2. The percentage of positive cells was quantified by FACS and normalized to the nontargeting control relative to the initial time for each sgRNA. Data from a representative experiment are shown. (C) Growth curve of MA9 Riok2fl/fl R-CreER cells transduced with lentiviruses expressing the indicated proteins. OHT was added to the cell culture medium at day 0 (n = 3 biological replicates). Multiple unpaired Student t tests were performed to assess statistical significance comparing RIOK2 WT and RIOK2K123A, D246A-expressing cell lines with the stuffer cell line. ***P < .001. ns, not significant. Error bars represent SD. (D) Diagram depicting RIOK2 phosphorylation sites based on data from PhosphoSitePlus.25 Asterisks mark phosphorylation sites mutated in the RIOK2 4A (red asterisks) and 8A (red and black asterisks) mutants. (E) Competition-based proliferation assays in murine MLL-AF9 cells expressing WT RIOK2, 4A or 8A mutants as indicated. The cells were transduced with a lentivirus expressing BFP and the indicated Riok2 sgRNAs. The percentage of positive cells was quantified by FACS and normalized to the nontargeting control relative to the initial time point for each sgRNA. Data from a representative experiment are shown.
The ATPase function of RIOK2 is essential for supporting leukemia proliferation. (A) Overview of the different RIOK2 mutants used for structure-function analysis. NES, nuclear export signal; wHTH, winged helix-turn-helix domain. (B) Competition-based proliferation assays in murine MLL-AF9 cells expressing the depicted RIOK2 mutants. Expression of luciferase (stuffer) was used as the negative control. MA9 cells were transduced with an sgRNA plasmid containing a BFP fluorophore to knock out endogenous Riok2. The percentage of positive cells was quantified by FACS and normalized to the nontargeting control relative to the initial time for each sgRNA. Data from a representative experiment are shown. (C) Growth curve of MA9 Riok2fl/fl R-CreER cells transduced with lentiviruses expressing the indicated proteins. OHT was added to the cell culture medium at day 0 (n = 3 biological replicates). Multiple unpaired Student t tests were performed to assess statistical significance comparing RIOK2 WT and RIOK2K123A, D246A-expressing cell lines with the stuffer cell line. ***P < .001. ns, not significant. Error bars represent SD. (D) Diagram depicting RIOK2 phosphorylation sites based on data from PhosphoSitePlus.25 Asterisks mark phosphorylation sites mutated in the RIOK2 4A (red asterisks) and 8A (red and black asterisks) mutants. (E) Competition-based proliferation assays in murine MLL-AF9 cells expressing WT RIOK2, 4A or 8A mutants as indicated. The cells were transduced with a lentivirus expressing BFP and the indicated Riok2 sgRNAs. The percentage of positive cells was quantified by FACS and normalized to the nontargeting control relative to the initial time point for each sgRNA. Data from a representative experiment are shown.
RIOK2 is an atypical kinase, and previous studies have suggested that because of the lack of substrate recognition domains, RIOK2 may not have the ability to phosphorylate other proteins.26 However, it has been reported that RIOK2 has the capability to autophosphorylate and that this activity is essential for its function.4,27 To test the requirement of RIOK2 autophosphorylation in AML, we decided to mutate all its potential autophosphorylation sites. Because it is not known which amino acid residues of human RIOK2 undergo autophosphorylation, we decided to mutate the sites with the highest abundance in phospho-mass spectrometry data sets and a site that has been described to be autophosphorylated in Archaeoglobus fulgidus (Figure 3D).25,27 Thus, we generated 2 RIOK2 mutants, in which 4 or 8 serine/threonine residues were mutated to alanines (supplemental Figure 2C-D). Because both mutants rescued the loss of Riok2 (Figure 3E), these results demonstrate that the phosphorylation of these 8 sites is not necessary for RIOK2 activity and for leukemic cell survival.
As RIOK2 is involved in the export of the 40S ribosomal subunit from the nucleus into the cytoplasm,4 we investigated whether the nuclear and/or cytoplasmic role of RIOK2 is important for leukemic cell survival. As suggested by the results presented in supplemental Figure 2E-G, both the cytosolic and the nuclear function of RIOK2 appear important for its ability to support cell survival.
Loss of RIOK2 leads to reduced protein synthesis and apoptosis in leukemic cells but not in fibroblasts
To understand the functional consequences of RIOK2 loss on AML proliferation in more detail, we first addressed the effects of Riok2 deletion on cell cycle progression. We performed EdU labeling followed by flow cytometry 6 days after sgRNA transduction. Loss of Riok2 led to a striking reduction of cells in S-phase and an increase in the apoptotic sub-G1 fraction (Figure 4A-B). Similar results were obtained in Riok2 R-Cre MA9 cell lines (supplemental Figure 3A), in which the decrease in proliferation and increase in apoptosis can be rescued by expression of WT RIOK2, but not the RIOK2K123A, D246A mutant.
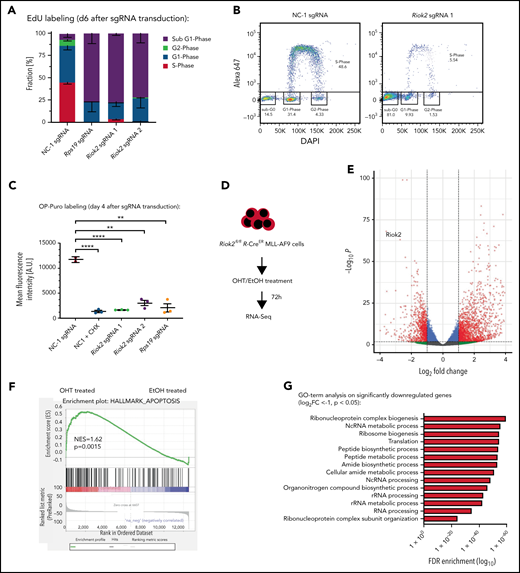
Loss of RIOK2 leads to decreased protein synthesis followed by apoptosis. (A) Quantification of FACS-based EdU labeling and calculation of cell cycle distribution of MA9 cells 6 days after transduction with lentiviruses expressing the indicated sgRNAs. n = 3 for each sgRNA. Error bars represent standard error of the mean. (B) Two representative FACS plots of EdU-labeled MA9 cells expressing either NC-1 or Riok2 sgRNAs. (C) OP-puro labeling followed by FACS analysis of cells transduced with lentiviruses expressing Riok2, Rps19, or NC-1 sgRNAs. The translation inhibitor cycloheximide was used as a positive control. Analysis was performed on FACS-sorted cells 4 days after sgRNA transduction. The Student t test was used to assess statistical significance. **P < .01; ****P < .0001. n = 2-3 biological replicates. Error bars represent SD. (D) Overview of the RNA-Seq experiments using MA9 Riok2fl/fl R-CreER treated with either OHT (n = 3 biological replicates) or EtOH (n = 3 biological replicates) for 72 hours. (E) Volcano plot showing upregulated and downregulated transcripts after loss of RIOK2. Significantly changed transcripts (P < .05; log2 FC > ±1) are shown in red. (F) Gene set enrichment analysis of apoptosis hallmark gene signatures comparing OHT vs EtOH treated Riok2fl/fl R-CreER MA9 cells. (G) Gene Ontology term analysis on significantly downregulated genes upon 72 of OHT treatments of Riok2fl/fl R-CreER MA9 cells (log2FC <−1; P < .05).
Loss of RIOK2 leads to decreased protein synthesis followed by apoptosis. (A) Quantification of FACS-based EdU labeling and calculation of cell cycle distribution of MA9 cells 6 days after transduction with lentiviruses expressing the indicated sgRNAs. n = 3 for each sgRNA. Error bars represent standard error of the mean. (B) Two representative FACS plots of EdU-labeled MA9 cells expressing either NC-1 or Riok2 sgRNAs. (C) OP-puro labeling followed by FACS analysis of cells transduced with lentiviruses expressing Riok2, Rps19, or NC-1 sgRNAs. The translation inhibitor cycloheximide was used as a positive control. Analysis was performed on FACS-sorted cells 4 days after sgRNA transduction. The Student t test was used to assess statistical significance. **P < .01; ****P < .0001. n = 2-3 biological replicates. Error bars represent SD. (D) Overview of the RNA-Seq experiments using MA9 Riok2fl/fl R-CreER treated with either OHT (n = 3 biological replicates) or EtOH (n = 3 biological replicates) for 72 hours. (E) Volcano plot showing upregulated and downregulated transcripts after loss of RIOK2. Significantly changed transcripts (P < .05; log2 FC > ±1) are shown in red. (F) Gene set enrichment analysis of apoptosis hallmark gene signatures comparing OHT vs EtOH treated Riok2fl/fl R-CreER MA9 cells. (G) Gene Ontology term analysis on significantly downregulated genes upon 72 of OHT treatments of Riok2fl/fl R-CreER MA9 cells (log2FC <−1; P < .05).
Considering the previously described role of RIOK2 in ribosome biogenesis, we hypothesized that RIOK2 loss could lead to cell death via decreasing protein synthesis through ribosome loss. To investigate the effects of RIOK2 loss on protein synthesis, we performed OP-puro labeling followed by fluorescence-activated cell-sorting analysis (FACS) 4 days after transduction of MA9 cells with sgRNAs. Treatment of MA9 cells with the translation inhibitor cycloheximide completely abolished translation, whereas loss of RIOK2 led to a strong reduction in translational output, similar to that observed in RPS19-depleted cells (Figure 4C). Accordingly, deletion of Riok2 in the MA9 Riok2 R-Cre cells led to a strong and significant downregulation of protein synthesis (supplemental Figure 3B).
These results were further supported by mass spectrometry experiments, in which we demonstrated that RIOK2 interacts with proteins of the translational machinery, including ribosomal proteins (supplemental Figure 3C-F). Moreover, gene set enrichment analysis on RNA-Seq data from Riok2fl/fl R-Cre MA9 cells showed a significant enrichment of the apoptotic hallmark gene set (Figure 4D-F) upon OHT treatment and the downregulation of genes associated with ribosome biogenesis, translation, and other biosynthetic processes (Figure 4G).
As we observed a differential dependency for RIOK2 between MLL-AF9 cells and MEFs in our initial analysis, we generated mouse embryonic fibroblasts from Riok2fl/fl R-CreERT and Riok2fl/+ CreERT mice (supplemental Figure 4A) to study the consequences of RIOK2 loss in a different cell type. The deletion of Riok2 in Riok2fl/fl MEFs led to growth arrest, whereas Riok2 heterozygous MEFs continued to proliferate (supplemental Figure 4B-C). Quantitative PCR analysis of senescence markers revealed a strong upregulation of Cdkn1a and a downregulation of Lmnb1, which are typical hallmarks of senescence (supplemental Figure 4D).28,29 We performed EdU labeling followed by FACS 6 days after Riok2 deletion and observed an increase of cells in G1-phase and a lower number of cells in S-phase (supplemental Figure 4E). Interestingly, we did not observe an increase in the number of cells undergoing apoptosis (supplemental Figure 4F). We also performed OP-puro labeling in Trp53−/− cells 4 days after transduction and observed no significant decrease in protein synthesis after RIOK2 loss, whereas knockout of Rps19 decreased protein synthesis similar that observed in the leukemic cells (supplemental Figure 4E).
In summary, we demonstrated that loss of RIOK2 leads to a significant downregulation of protein synthesis, followed by cell cycle arrest and apoptosis in AML. Intriguingly, the short-term effects of losing RIOK2 were considerably milder in MEFs.
RIOK2 loss leads to translational stalling followed by ribosome degradation in leukemic cells
Based on the downregulation of protein synthesis after RIOK2 loss and the previously described role of RIOK2 in 40S subunit maturation, we wondered whether this phenotype is a consequence of impaired ribosome biogenesis. To investigate the consequences of RIOK2 loss on the formation of mature ribosomes, we performed polysome profiling 40 and 72 hours after addition of OHT or ethanol in Riok2fl/fl R-Cre cells (Figure 5A). We also included short-term (12 hours) treatment with 200 nM doxorubicin as a positive control (Figure 5B), as it has been shown to cause translational stalling and ribosomal degradation.30 RIOK2 loss led to a dramatic reduction in polysomes compared with WT cells and an increase in mature 80S ribosomes (Figure 5A), similar to that observed with doxorubicin treatment (Figure 5B), whereas only minor differences were observed for the 40S peak. This finding suggests that loss of RIOK2 initially leads to ribosomal stalling instead of impaired ribosome biogenesis in leukemic cells. Longer term absence of RIOK2 led to a complete loss of the polysome population (Figure 5C). In addition, the 80S monosomes and the 40S and 60S subunits were not present in detectable levels (Figure 5C). Consistent with this result, proteins from the small (RPS3a, RPS2) and large (RPL22) ribosomal subunits were strongly reduced in RIOK2-depleted cells (Figure 5D). Taken together, these results extend previous observations4 and show that RIOK2 is essential for ribosome stability in AML.
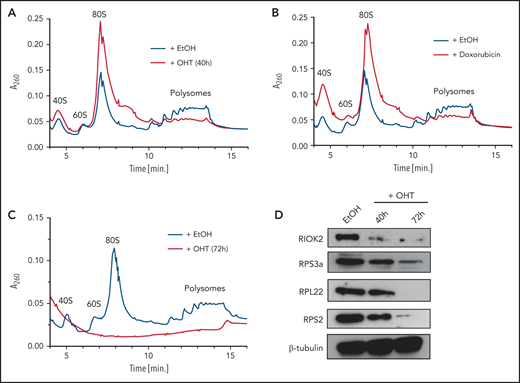
RIOK2 loss leads to ribosomal stalling and ribosome degradation. (A) Polysome profiles of lysates prepared from MA9 Riok2fl/fl R-CreER cells treated for 40 hours with EtOH or OHT. (B) Polysome profiles of lysates prepared from MA9 Riok2fl/fl R-CreER cells treated for 40 hours with EtOH or with 200 nM doxorubicin for 12 hours. EtOH control is the same sample as in panel A. (C) Polysome profiles of lysates prepared from MA9 Riok2fl/fl R-CreER cells treated with EtOH or OHT for 72 hours. (D) Western blot analysis for the indicated proteins of MA9 Riok2fl/fl R-CreER cells treated with EtOH for 72 hours or with OHT for 40 or 72 hours, as indicated.
RIOK2 loss leads to ribosomal stalling and ribosome degradation. (A) Polysome profiles of lysates prepared from MA9 Riok2fl/fl R-CreER cells treated for 40 hours with EtOH or OHT. (B) Polysome profiles of lysates prepared from MA9 Riok2fl/fl R-CreER cells treated for 40 hours with EtOH or with 200 nM doxorubicin for 12 hours. EtOH control is the same sample as in panel A. (C) Polysome profiles of lysates prepared from MA9 Riok2fl/fl R-CreER cells treated with EtOH or OHT for 72 hours. (D) Western blot analysis for the indicated proteins of MA9 Riok2fl/fl R-CreER cells treated with EtOH for 72 hours or with OHT for 40 or 72 hours, as indicated.
Pharmacological inhibition of RIOK2 has antileukemic effects in vitro and in vivo
To obtain further experimental proof of concept for targeting RIOK2 in AML, we used a recently described small-molecule inhibitor of RIOK2 (1-[2-(2-thiazolyl)diazenyl]-2-naphthalenol, hereafter referred to as RIOK2i; Figure 6A).13 The RIOK2 inhibitor efficiently inhibited the proliferation of the tested mouse and human AML cell lines and, less efficiently, the mouse and human diploid fibroblasts (Figure 6B-C). This observation is in line with and extends the differential phenotypic effects observed by RIOK2 loss in fibroblasts and leukemic cells (Figure 1C).
![Pharmacological inhibition of RIOK2 has antileukemic effects in vivo and in vitro. (A) Chemical structure of RIOK2i (1-[2-(2-thiazolyl)diazenyl]-2-naphthalenol). (B) Viability of MA9 andTrp53−/− MEFs, after 72 hours of incubation with RIOK2i at the indicated concentrations. The data points show the mean ± SD of 3 technical replicates; 50% effective concentrations (EC50) were calculated with GraphPad Prism. (C) Viability of BJ-hTERT, MOLM13, and THP-1 cells after 72 hours of incubation with RIOK2i at the indicated concentrations. The data points show the mean ± SD of 3 technical replicates. EC50 values were calculated with GraphPad Prism. (D) Growth curves of MA9 cells treated as indicated over the course of 4 days. The data points show the mean ± SD. Multiple unpaired Student t tests were performed to assess the statistical significance of the comparison of RIOK2i treatment conditions with the DMSO-treated condition. ***P < .001. The experiment was performed in 3 biological replicates. (E) OP-puro labeling of MLL-AF9 (left) and MOLM13 cells (right) treated as indicated for 48 hours. A Student t test was used to assess statistical significance. *P < .05; **P < .01. n = 3 technical replicates. (F) Quantification of FACS-based EdU labeling and estimation of cell cycle profiles of murine MA9 or MOLM13 cells treated with DMSO or 300 nM or 1 µM RIOK2i for 72 hours. Error bars represent standard error of the mean. (G) Experimental overview of RIOK2i in vivo treatment strategy. SJL-B6 mice were sublethally irradiated and injected with 50 000 GFP-MA9 Cas9 cells the following day. FACS analysis to validate engraftment of leukemic cells was performed after 14 days. After validation, mice were treated with RIOK2i for 3 days per week at a concentration of 120 mg/kg. FACS analysis was performed at days 21 and 25 to check for the presence of leukemic cells. (H) FACS-based quantification of GFP-positive leukemic cells in erythrocyte depleted peripheral blood (PB) comparing oil-treated with RIOK2i-treated mice at the indicated time points. The Student t test was used to assess statistical significance. *P < .05. ns, not significant. (I) Model explaining the functional consequences of RIOK2 loss on protein synthesis and ribosomal stability in leukemic cells.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/139/2/10.1182_blood.2021012629/3/m_bloodbld2021012629f6.png?Expires=1768929903&Signature=q-9NtYu6Xfa9qkh5annhZiYq7YA6x0utSuUpwGE0LTOXPH2Tkykqz2PE9Dtze4rHGcv5eK4v5lZDa6bxmpZCphTiwGYiWya88W7xqngkvf2hsGE3tXW2SGDMoi--Hp3bMEgvt7ctMg9FILecLhj1l3hcrdusvH8IKMqb0wg0ZmtMHiiLCwtHyvphtpSw-Eu1B3QHElDy4~haG2MdDAzTGaZEHq8pf89M6jmGUOCgxZSICYgvhBK2FH5pEsMJZQFFEJIUWV950NRpr6bJCIpYe4LmaAquWorJQamf7YybihBld-pL0TZnRShgXXrC7Ofo5VN9GhqWWdKAwPVfw1fRyw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Pharmacological inhibition of RIOK2 has antileukemic effects in vivo and in vitro. (A) Chemical structure of RIOK2i (1-[2-(2-thiazolyl)diazenyl]-2-naphthalenol). (B) Viability of MA9 andTrp53−/− MEFs, after 72 hours of incubation with RIOK2i at the indicated concentrations. The data points show the mean ± SD of 3 technical replicates; 50% effective concentrations (EC50) were calculated with GraphPad Prism. (C) Viability of BJ-hTERT, MOLM13, and THP-1 cells after 72 hours of incubation with RIOK2i at the indicated concentrations. The data points show the mean ± SD of 3 technical replicates. EC50 values were calculated with GraphPad Prism. (D) Growth curves of MA9 cells treated as indicated over the course of 4 days. The data points show the mean ± SD. Multiple unpaired Student t tests were performed to assess the statistical significance of the comparison of RIOK2i treatment conditions with the DMSO-treated condition. ***P < .001. The experiment was performed in 3 biological replicates. (E) OP-puro labeling of MLL-AF9 (left) and MOLM13 cells (right) treated as indicated for 48 hours. A Student t test was used to assess statistical significance. *P < .05; **P < .01. n = 3 technical replicates. (F) Quantification of FACS-based EdU labeling and estimation of cell cycle profiles of murine MA9 or MOLM13 cells treated with DMSO or 300 nM or 1 µM RIOK2i for 72 hours. Error bars represent standard error of the mean. (G) Experimental overview of RIOK2i in vivo treatment strategy. SJL-B6 mice were sublethally irradiated and injected with 50 000 GFP-MA9 Cas9 cells the following day. FACS analysis to validate engraftment of leukemic cells was performed after 14 days. After validation, mice were treated with RIOK2i for 3 days per week at a concentration of 120 mg/kg. FACS analysis was performed at days 21 and 25 to check for the presence of leukemic cells. (H) FACS-based quantification of GFP-positive leukemic cells in erythrocyte depleted peripheral blood (PB) comparing oil-treated with RIOK2i-treated mice at the indicated time points. The Student t test was used to assess statistical significance. *P < .05. ns, not significant. (I) Model explaining the functional consequences of RIOK2 loss on protein synthesis and ribosomal stability in leukemic cells.
Pharmacological inhibition of RIOK2 has antileukemic effects in vivo and in vitro. (A) Chemical structure of RIOK2i (1-[2-(2-thiazolyl)diazenyl]-2-naphthalenol). (B) Viability of MA9 andTrp53−/− MEFs, after 72 hours of incubation with RIOK2i at the indicated concentrations. The data points show the mean ± SD of 3 technical replicates; 50% effective concentrations (EC50) were calculated with GraphPad Prism. (C) Viability of BJ-hTERT, MOLM13, and THP-1 cells after 72 hours of incubation with RIOK2i at the indicated concentrations. The data points show the mean ± SD of 3 technical replicates. EC50 values were calculated with GraphPad Prism. (D) Growth curves of MA9 cells treated as indicated over the course of 4 days. The data points show the mean ± SD. Multiple unpaired Student t tests were performed to assess the statistical significance of the comparison of RIOK2i treatment conditions with the DMSO-treated condition. ***P < .001. The experiment was performed in 3 biological replicates. (E) OP-puro labeling of MLL-AF9 (left) and MOLM13 cells (right) treated as indicated for 48 hours. A Student t test was used to assess statistical significance. *P < .05; **P < .01. n = 3 technical replicates. (F) Quantification of FACS-based EdU labeling and estimation of cell cycle profiles of murine MA9 or MOLM13 cells treated with DMSO or 300 nM or 1 µM RIOK2i for 72 hours. Error bars represent standard error of the mean. (G) Experimental overview of RIOK2i in vivo treatment strategy. SJL-B6 mice were sublethally irradiated and injected with 50 000 GFP-MA9 Cas9 cells the following day. FACS analysis to validate engraftment of leukemic cells was performed after 14 days. After validation, mice were treated with RIOK2i for 3 days per week at a concentration of 120 mg/kg. FACS analysis was performed at days 21 and 25 to check for the presence of leukemic cells. (H) FACS-based quantification of GFP-positive leukemic cells in erythrocyte depleted peripheral blood (PB) comparing oil-treated with RIOK2i-treated mice at the indicated time points. The Student t test was used to assess statistical significance. *P < .05. ns, not significant. (I) Model explaining the functional consequences of RIOK2 loss on protein synthesis and ribosomal stability in leukemic cells.
To test whether the mechanism of action of the RIOK2 inhibitor was consistent with an on-target effect, we performed OP-puro as well as EdU labeling and polysome profiling after RIOK2i treatment. These experiments showed that the RIOK2 inhibitor affected protein synthesis, ribosome profiles, and cell cycle distribution that were comparable to the effects observed by deleting Riok2 (Figure 6D-F; supplemental Figure 5A). Further support for the on-target activity of the RIOK2 inhibitor was obtained by measuring its effect on gene expression. Although the number of genes with differential expression in response to the RIOK2 inhibitor was substantially lower than those observed by deleting Riok2, there was a statistically significant overlap between the changes in gene expression (supplemental Figure 5B). Taken together, these results support the previous observations that the RIOK2 inhibitor is specific13 and that the effects observed in the AML cell lines are caused by RIOK2 inhibition.
The RIOK2 inhibitor has been used for treatment of a prostate cancer xenograft mouse model in vivo.13 In the concentrations used (100-150 mg/kg) the inhibitor was shown to be well tolerated in nude mice and led to inhibition of tumor growth.13 To obtain further proof of concept for targeting RIOK2 in AML, we proceeded to assess the efficacy of using RIOK2i in vivo. We transplanted MA9 cells into sublethally irradiated B6-SJL mice and treated the mice by injecting the compound at a dose of 120 mg/kg 3 times per week (Figure 6G). Consistent with previously published data,13 we did not observe adverse effects on blood composition or body weight after treatment of mice using this regimen (supplemental Figure 5C-D). To assess the efficacy of RIOK2 inhibition in vivo, we measured protein synthesis by in vivo OP-puro labeling of nucleated cells in the peripheral blood 1, 4, and 24 hours after RIOK2i injection. The RIOK2i treatment led to a significant reduction in protein synthesis 1 and 4 hours after injection, whereas we observed a significant rebound of protein synthesis 24 hour after injection (supplemental Figure 5E). This result suggests that the RIOK2 inhibitor engages its target in vivo; however, the inhibitor is not sufficiently stable to maintain RIOK2 inhibition with the dose used for 24 hours. Despite this finding, we observed a significant reduction in the number of leukemic cells at both days 21 and 25 after transplantation in RIOK2i-treated mice vs the control (Figure 6H). However, this treatment regimen led to a mild, but not significant, increase in overall survival of the transplanted recipient mice (supplemental Figure 5F). We tested higher concentrations of the RIOK2 inhibitor for the treatments of the mice; however, the experiment had to be stopped because the treated mice lost weight (data not shown). Taken together, the results showed that pharmacological targeting of RIOK2 ATPase activity inhibits leukemic cell proliferation in vivo and in vitro.
Discussion
Based on our results, we suggest a novel strategy to target protein synthesis in AML by inhibiting RIOK2 ATPase activity. Loss of RIOK2 causes a reduction of protein synthesis by ribosome degradation in leukemic cells, but not in fibroblasts. Despite ribosome biogenesis and maintenance being essential cellular processes, inhibition of ribosome biogenesis has been proposed as a potential therapeutic strategy in cancer.3 Recently, the RNA-Pol I transcription inhibitor CX-5461 has shown clinical benefits in advanced hematological cancers in a phase 1 dose-escalation study and is currently being tested in several phase 1 clinical trials.31 The mechanism of action of CX-5461 relies on WT p53, as the accumulation of free ribosomal proteins activates a p53-mediated stress response.32 In contrast, RIOK2 inhibition has antileukemic effects in the absence of p53, thereby potentially expanding the use of inhibitors targeting protein synthesis to p53 mutant cancers. In addition, targeting protein synthesis through RIOK2 inhibition could act as an alternative to current approaches targeting the PI3K/AKT/mTOR pathway, which, despite its significance in AML pathogenesis, have not shown clinical effectiveness in AML treatment.12
We have shown that loss of RIOK2 activity, by either genetic deletion or small-molecule inhibition, leads to a decrease of actively translated mRNAs followed by ribosome degradation (Figure 6I). Interestingly, leukemic cells are more sensitive to RIOK2 inhibition than fibroblasts. Impaired ribosome biogenesis, caused, for example, by haploinsufficiency of ribosomal genes or defects in ribosome biogenesis factors, causes a series of disorders called ribosomopathies, which often exhibit tissue-specific defects.33 Similarly, differential effect for RIOK2 knockdown have been observed for glioblastoma cells and noncancerous glial cells. Although the reasons for the differential RIOK2 dependency between the various cell types is unknown, we speculate that because of a frequent disruption of mechanisms that regulate protein synthesis in cancer cells, these cells undergo apoptosis related to an inability to adequately compensate for the sudden loss of ribosome homeostasis.
We have shown that pharmacological inhibition of RIOK2 leads to significantly decreased growth of leukemic cells in vivo without having adverse effects on body weight or blood composition. This finding is in agreement with published results showing that RIOK2 inhibition decreases the growth of prostate cancer cells in vivo.13 However, with the dosing regimen used in our study, the RIOK2 inhibitor reduced protein synthesis in vivo for only a short time, and consistent with that result, the compound led to a slight, but not significant, increase in the survival of the leukemic mice. Thus, pharmacological optimization of compound stability or other dosing schedules is necessary for its clinical use.
Although further investigation into the role of RIOK2 in normal hematopoiesis and the development of pharmacologically improved inhibitors targeting RIOK2 is needed to evaluate the suitability of RIOK2 as a target in AML, we present a novel approach targeting protein synthesis in cancer by affecting ribosome stability that has the potential to expand the current portfolio of treatment strategies for AML.
Acknowledgments
The authors thank members of the K.H. group for discussions; the Danish Technical University Proteomics Core Facility for collaboration in performing the mass spectrometry experiments; and the FACS Core Facility at the Biotech Research and Innovation Centre for assistance with the FACS experiments.
J.-E.M. was supported by the Novo Nordisk Foundation (NNF) Copenhagen Bioscience Program (NNF18CC0033666). The work in the K.H. laboratory was supported by a the Danish Cancer Society (R167-A10877), by The Neye Foundation, by a center grant (NNF17CC0027852) from the NNF to the NNF Center for Stem Cell Biology, and by National Institutes of Health, National Cancer Institute Support Grant P30 CA008748 to the Memorial Sloan Kettering Cancer Center.
Authorship
Contribution: J.-E.M., K.A., and K.H. designed the experiments and analyzed the data; J.-E.M., K.A., H.M.K., and K.K. performed the experiments; K.L.A., supervised by A.H.L., performed the polysome profiling experiments and analyzed the data; and J.-E.M. and K.H. wrote the manuscript.
Conflict-of-interest disclosure: K.H. is a consultant for Inthera Bioscience AG and a scientific advisor for MetaboMed Inc and Hannibal Health Innovation.
Correspondence: Kristian Helin, The Institute of Cancer Research, 237 Fulham Rd, London SW3 6JB, United Kingdom; e-mail: kristian.helin@icr.ac.uk.
RNA-Seq data have been deposited in the Gene Expression Omnibus repository (accession number GSE174859).
Original experimental data concerning the publication are available by e-mail request to the corresponding author (helink@mskcc.org).
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.