

Key Points
ALK-positive histiocytosis is a distinct entity associated with KIF5B-ALK fusions and characterized by frequent neurologic involvement.
ALK inhibition induces robust and durable responses in patients with ALK-positive histiocytosis.

Abstract
ALK-positive histiocytosis is a rare subtype of histiocytic neoplasm first described in 2008 in 3 infants with multisystemic disease involving the liver and hematopoietic system. This entity has subsequently been documented in case reports and series to occupy a wider clinicopathologic spectrum with recurrent KIF5B-ALK fusions. The full clinicopathologic and molecular spectra of ALK-positive histiocytosis remain, however, poorly characterized. Here, we describe the largest study of ALK-positive histiocytosis to date, with detailed clinicopathologic data of 39 cases, including 37 cases with confirmed ALK rearrangements. The clinical spectrum comprised distinct clinical phenotypic groups: infants with multisystemic disease with liver and hematopoietic involvement, as originally described (Group 1A: 6/39), other patients with multisystemic disease (Group 1B: 10/39), and patients with single-system disease (Group 2: 23/39). Nineteen patients of the entire cohort (49%) had neurologic involvement (7 and 12 from Groups 1B and 2, respectively). Histology included classic xanthogranuloma features in almost one-third of cases, whereas the majority displayed a more densely cellular, monomorphic appearance without lipidized histiocytes but sometimes more spindled or epithelioid morphology. Neoplastic histiocytes were positive for macrophage markers and often conferred strong expression of phosphorylated extracellular signal-regulated kinase, confirming MAPK pathway activation. KIF5B-ALK fusions were detected in 27 patients, whereas CLTC-ALK, TPM3-ALK, TFG-ALK, EML4-ALK, and DCTN1-ALK fusions were identified in single cases. Robust and durable responses were observed in 11/11 patients treated with ALK inhibition, 10 with neurologic involvement. This study presents the existing clinicopathologic and molecular landscape of ALK-positive histiocytosis and provides guidance for the clinical management of this emerging histiocytic entity.
Introduction
Histiocytic disorders are a group of rare diseases characterized by the accumulation of macrophage-, dendritic cell–, or monocyte-differentiated cells in various tissues and organs.1,2 With the advent of molecular technologies, recurrent genetic alterations have been identified in several histiocytoses,3-17 reframing the conceptualization of these disorders from one of primary inflammatory conditions to that of clonal neoplastic diseases.18,19 These alterations primarily affect genes encoding protein kinases of the intracellular mitogen-activated protein kinase (MAPK) signaling pathway, leading to constitutive activation of this pathway (Figure 1).11 However, downstream extracellular signal-regulated kinase (ERK) activation has also been observed in histiocytoses without detected MAPK pathway mutations,3,11,13 leading to the notion that histiocytic neoplasms are uniformly characterized by dependence on MAPK signaling.20 This has led to the clinical implementation of pharmacological inhibitors of the MAPK signaling pathway (BRAF and/or MEK inhibitors) for the treatment of patients with severe and/or relapsed/refractory histiocytic disease.20-27 In addition to MAPK pathway mutations, recurrent activating genetic alterations in genes encoding protein kinases of the PI3K/AKT/mTOR signaling pathway (ie, PIK3CA) or receptor tyrosine kinases (ie, CSF1R, NTRK1, RET, or ALK) have been identified in a subset of histiocytic neoplasms.6,13 Furthermore, significant associations between specific kinase alterations and clinicopathologic phenotypes have recently been observed.6 These findings pave the way for molecular (sub)classification of histiocytic neoplasms,1,6,8,28 and highlight the potential for targeted therapy beyond BRAF or MEK inhibition in histiocytosis patients.

Schematic overview of downstream ALK signaling through MAPK and PI3K/AKT/mTOR signaling pathways. ALK is a classical receptor tyrosine kinase consisting of an extracellular ligand-binding domain, a transmembrane domain, and an intracellular tyrosine kinase domain.76 In ALK fusions such as KIF5B-ALK, the amino-terminal fusion partner is fused to the intracellular tyrosine kinase domain of ALK, leading to constitutive activation of downstream signaling, including RAS-RAF-MEK-ERK (MAPK) and PI3K/AKT/mTOR signaling pathways. MAPK pathway activation ultimately leads to phosphorylation of downstream ERK, which can enter the nucleus and increase the transcription of various effector genes, including the gene encoding for Cyclin D1 (CCND1). Translation of CCND1 messenger RNA to the Cyclin D1 protein is mTOR-dependent.77 Figure adapted from Emile et al,2 with permission from the authors.
Schematic overview of downstream ALK signaling through MAPK and PI3K/AKT/mTOR signaling pathways. ALK is a classical receptor tyrosine kinase consisting of an extracellular ligand-binding domain, a transmembrane domain, and an intracellular tyrosine kinase domain.76 In ALK fusions such as KIF5B-ALK, the amino-terminal fusion partner is fused to the intracellular tyrosine kinase domain of ALK, leading to constitutive activation of downstream signaling, including RAS-RAF-MEK-ERK (MAPK) and PI3K/AKT/mTOR signaling pathways. MAPK pathway activation ultimately leads to phosphorylation of downstream ERK, which can enter the nucleus and increase the transcription of various effector genes, including the gene encoding for Cyclin D1 (CCND1). Translation of CCND1 messenger RNA to the Cyclin D1 protein is mTOR-dependent.77 Figure adapted from Emile et al,2 with permission from the authors.
In 2008, Chan et al first described 3 infants with a novel type of systemic histiocytosis characterized by ALK immunoreactivity in large CD163+ histiocytes with variable expression of other histiocyte/dendritic cell markers (S100, fascin, factor XIIIa).29 Clinically, the disease was characterized by liver and hematopoietic involvement and could be confused with a storage disorder or leukemia. The disease tended to resolve slowly, sometimes even without any specific treatment but only supportive care. One case demonstrated a TPM3-ALK translocation. Over the following years, several individual case reports and small case series about ALK+ histiocytosis have been published,30-49 confirming the rare but indefinite occurrence of this emerging histiocytic entity. Most notably, Chang et al described a series of 10 cases,43 including the original 3 cases reported by Chan et al, with 8 having molecular confirmation of ALK rearrangements. Together, these reports have documented ALK-positive histiocytosis in both older children and adults with single- or multisystemic disease, expanding beyond the initial observation of the disease as a systemic disorder of infancy. Moreover, the frequent presence of KIF5B-ALK fusions has been reported,43 and successful treatment of some patients with ALK inhibition has been described.6,27,33,34,39,40,43,50,51 Due to the modest number of reported patients, and particularly few cases with clinical information and/or successful molecular analysis, the full clinicopathologic and molecular spectra of ALK-positive histiocytosis remain, however, incompletely characterized. Accordingly, here we describe the results of the largest study of ALK-positive histiocytosis to date, outlining the clinicopathologic features of 39 patients, including 37 cases with proven ALK rearrangements.
Materials and methods
Patient selection and confirmation of diagnosis
Fifty-two cases from different hospitals throughout North America, Europe, and Australia were compiled for this international retrospective study. Six cases were previously published6,30,31,39,44,47 and are reported with updated follow-up (when available). All cases underwent central review of pathology slides (J.P. and J.-F.E.) for confirmation of diagnosis. Additional immunohistochemical and/or molecular analyses were performed when deemed appropriate. Criteria for a diagnosis of ALK-positive histiocytosis and inclusion in the study were the following: (1) histologic confirmation of a histiocytosis,1 with expression of at least 2 macrophage/histiocyte markers (including CD163, CD68, CD14, and/or CD4) by the lesional cells and (2A) molecular confirmation of ALK rearrangement and/or (2B) classic infantile systemic disease with diffuse strong ALK immunoreactivity as previously described.29 Cases with ALK rearrangements in which immunohistochemistry revealed histiocytes to be disparate from lesional ALK+ cells without immunoreactivity for macrophage/histiocyte markers were termed “atypical ALK-rearranged histiocyte-rich tumors” and not included in the primary study cohort. Also, histiocytosis cases with ALK immunoreactivity but without ALK rearrangements by comprehensive molecular analysis (ie, RNA sequencing [RNA-seq]) were not included in the primary study cohort. Cases excluded for these 2 reasons are characterized separately. Written informed consent for this study was obtained from the patients and/or their legal representative when required, or a waiver of consent was obtained from the relevant institutional review board for retrospective research.
Clinical and radiologic assessment and data collection
Clinical information was extracted from the medical records by the treating physicians and provided in a pseudonymized fashion according to a standardized deidentified case report form. Retrieved data included demographic characteristics, presenting symptoms, sites of disease involvement, treatments, and treatment outcomes. First- and second-or-further line treatments were categorized as observation with supportive care, surgical resection, radiotherapy, conventional systemic therapy (ie, cytotoxic chemotherapy, corticosteroids, immunosuppression, and/or interferon-α), ALK inhibition, or a combination of these. As previously published for Erdheim-Chester disease (ECD) and Langerhans cell histiocytosis (LCH),27,52,53 responses to treatment as documented in the medical record were classified as complete response (complete resolution of disease), partial response (partial resolution of disease), stable disease (no significant change in lesions), or progressive disease (growth of known lesions or appearance of new lesions). Responses were extracted from the clinical medical records and based on formal clinical interpretations of ultrasound, computed tomography (CT), magnetic resonance imaging (MRI) and/or positron emission tomography (PET)-CT when available. The less favorable radiologic response was prioritized if multiple imaging methods were used that showed discrepant responses. Objective response was defined as partial or complete response. Whether or not progression or relapse of disease was subsequently observed was dichotomously captured. Progression was defined as progressive disease or death from any cause.
Histopathologic analysis
Histopathologic information was collected in a standardized deidentified case report form. Morphology and immunophenotype were reviewed for all cases (J.P. and J.-F.E.). Immunohistochemistry (IHC) was performed as part of initial diagnosis at participating institutions and consequently was executed using varying staining techniques and monoclonal antibodies. If ALK, CD163, CD68, S100, and/or CD1a IHC was not originally performed, these stains were done in the process of central review when sufficient tumor tissue was available. The ALK clones that were used for each case are shown in supplemental Table 1, available on the Blood Web site. The phosphorylated-ERK (p-ERK) immunostains were all performed using clone D13.14.4E (Cell Signaling).
Molecular pathologic analysis
Molecular pathologic information was collected in a standardized deidentified case report form. Molecular analysis was performed as part of initial diagnosis at referring institutions or done in the process of central review, when sufficient tumor tissue was available. For analyses performed during central review, fluorescence in situ hybridization (FISH) analysis was performed using CytoCell dual color ALK break-apart probes (Oxford Gene Technology) as previously described.54 Targeted RNA-seq was performed using customized Archer FusionPlex next-generation sequencing (NGS) panels, which allow the detection of both fusions with known or unknown fusion partners. For NGS analysis at the European referral center Ambroise Paré Hospital (Paris, France), RNA was isolated using the Maxwell Rapid Sample Concentrator RNA Formalin-Fixed Paraffin-Embedded Kit (Promega), and RNA concentrations were measured using the Qubit RNA High Sensitivity Assay Kit (Thermo Fisher Scientific). Successful NGS required 20 to 250 ng RNA per reaction. Generated libraries were sequenced using a MiSeq system (Illumina), and produced reads were analyzed with Archer Analysis software. Archer RNA NGS at the North American referral center Memorial Sloan-Kettering Cancer Center (New York, NY) was performed as previously described.55 Clonality assessment of T-cell receptor or immunoglobulin (Ig) gene rearrangements was performed using standardized multiplex polymerase chain reaction assays as previously described.56,57
Statistical analysis
Data were analyzed using descriptive statistics. Continuous variables were summarized with medians and ranges, and categorical variables were summarized with frequencies and proportions. Frequency of objective responses were summarized for treatment modality by patient group and for the entire cohort, and the frequency of disease progression or relapse following therapy was summarized.
Results
Thirty-nine patients meeting criteria for ALK-positive histiocytosis were identified and included in the study (Tables 1-3; supplemental Tables 1-3). In addition, 3 patients with atypical ALK-rearranged histiocyte-rich tumors were recognized (Table 3), and 10 patients were identified with histiocytoses demonstrating ALK immunoreactivity but no ALK rearrangement by RNA-seq (supplemental Table 4). These 13 patients without complete criteria for ALK-positive histiocytosis diagnosis were excluded from the primary study cohort and are described separately.
Clinical and molecular characteristics of the distinct clinical phenotypic groups of ALK-positive histiocytosis and the overall cohort
| Group 1A (n = 6) | Group 1B (n = 10) | Group 2 (n = 23) | Overall (n = 39) | |
|---|---|---|---|---|
| Age at presentation | ||||
| Median age (range) | 1.5 mo (0-5 mo) | 14.5 y (0-41 y) | 7 y (0-41 y) | 3 y (0-41 y) |
| Child | 6 (100%) | 5 (50%) | 20 (87%) | 31 (79%) |
| Adult | 5 (50%) | 3 (13%) | 8 (21%) | |
| Sex | ||||
| Female | 4 (67%) | 7 (70%) | 13 (57%) | 24 (62%) |
| Male | 2 (33%) | 3 (30%) | 10 (43%) | 15 (38%) |
| Organ involvement | ||||
| Nervous system | 7 (70%) | 12 (52%) | 19 (49%) | |
| Liver | 6 (100%) | 5 (50%) | 11 (28%) | |
| Lung | 1 (17%) | 7 (70%) | 1 (4%) | 9 (23%) |
| Bone | 7 (70%) | 2 (9%) | 9 (23%) | |
| Skin | 1 (17%) | 4 (40%) | 4 (17%) | 9 (23%) |
| Soft tissue | 3 (30%) | 4 (17%) | 7 (18%) | |
| Hematopoietic system | 6 (100%) | 6 (15%) | ||
| Spleen | 5 (83%) | 5 (13%) | ||
| Kidney | 2 (33%) | 2 (20%) | 4 (10%) | |
| Lymph node | 4 (40%) | 4 (10%) | ||
| Breast | 2 (20%) | 2 (5%) | ||
| Pancreas | 2 (20%) | 2 (5%) | ||
| Other* | 2 (20%) | 2 (5%) | ||
| ALK rearrangement | ||||
| KIF5B-ALK | 1 (17%) | 7 (70%) | 19 (83%) | 27 (69%) |
| CLTC-ALK | 1 (17%) | 1 (3%) | ||
| TPM3-ALK | 1 (10%) | 1 (3%) | ||
| TFG-ALK | 1 (10%) | 1 (3%) | ||
| EML4-ALK | 1 (4%) | 1 (3%) | ||
| DCTN1-ALK | 1 (4%) | 1 (3%) | ||
| ALK-FISH+ | 2 (33%) | 1 (10%) | 2 (9%) | 5 (13%) |
| Not confirmed | 2 (33%) | 2 (5%) | ||
| Median follow-up (range) | 3.75 y (0-7 y) | 2 y (0-6 y) | 14 mo (0-18 y) | 21 mo (0-18 y) |
| Status at last follow-up | ||||
| Alive with | ||||
| no evidence of disease | 4 (67%) | 5 (50%) | 14 (61%) | 23 (59%) |
| regressive disease | 4 (40%) | 5 (22%) | 9 (23%) | |
| stable disease | 1 (4%) | 1 (3%) | ||
| recent diagnosis | 1 (4%) | 1 (3%) | ||
| Dead | 1 (17%) | 1 (10%) | 2 (5%) | |
| Lost to follow-up | 1 (17%) | 2 (9%) | 3 (8%) |
| Group 1A (n = 6) | Group 1B (n = 10) | Group 2 (n = 23) | Overall (n = 39) | |
|---|---|---|---|---|
| Age at presentation | ||||
| Median age (range) | 1.5 mo (0-5 mo) | 14.5 y (0-41 y) | 7 y (0-41 y) | 3 y (0-41 y) |
| Child | 6 (100%) | 5 (50%) | 20 (87%) | 31 (79%) |
| Adult | 5 (50%) | 3 (13%) | 8 (21%) | |
| Sex | ||||
| Female | 4 (67%) | 7 (70%) | 13 (57%) | 24 (62%) |
| Male | 2 (33%) | 3 (30%) | 10 (43%) | 15 (38%) |
| Organ involvement | ||||
| Nervous system | 7 (70%) | 12 (52%) | 19 (49%) | |
| Liver | 6 (100%) | 5 (50%) | 11 (28%) | |
| Lung | 1 (17%) | 7 (70%) | 1 (4%) | 9 (23%) |
| Bone | 7 (70%) | 2 (9%) | 9 (23%) | |
| Skin | 1 (17%) | 4 (40%) | 4 (17%) | 9 (23%) |
| Soft tissue | 3 (30%) | 4 (17%) | 7 (18%) | |
| Hematopoietic system | 6 (100%) | 6 (15%) | ||
| Spleen | 5 (83%) | 5 (13%) | ||
| Kidney | 2 (33%) | 2 (20%) | 4 (10%) | |
| Lymph node | 4 (40%) | 4 (10%) | ||
| Breast | 2 (20%) | 2 (5%) | ||
| Pancreas | 2 (20%) | 2 (5%) | ||
| Other* | 2 (20%) | 2 (5%) | ||
| ALK rearrangement | ||||
| KIF5B-ALK | 1 (17%) | 7 (70%) | 19 (83%) | 27 (69%) |
| CLTC-ALK | 1 (17%) | 1 (3%) | ||
| TPM3-ALK | 1 (10%) | 1 (3%) | ||
| TFG-ALK | 1 (10%) | 1 (3%) | ||
| EML4-ALK | 1 (4%) | 1 (3%) | ||
| DCTN1-ALK | 1 (4%) | 1 (3%) | ||
| ALK-FISH+ | 2 (33%) | 1 (10%) | 2 (9%) | 5 (13%) |
| Not confirmed | 2 (33%) | 2 (5%) | ||
| Median follow-up (range) | 3.75 y (0-7 y) | 2 y (0-6 y) | 14 mo (0-18 y) | 21 mo (0-18 y) |
| Status at last follow-up | ||||
| Alive with | ||||
| no evidence of disease | 4 (67%) | 5 (50%) | 14 (61%) | 23 (59%) |
| regressive disease | 4 (40%) | 5 (22%) | 9 (23%) | |
| stable disease | 1 (4%) | 1 (3%) | ||
| recent diagnosis | 1 (4%) | 1 (3%) | ||
| Dead | 1 (17%) | 1 (10%) | 2 (5%) | |
| Lost to follow-up | 1 (17%) | 2 (9%) | 3 (8%) |
Cervix, thyroid, salivary glands, and colorectum.
Clinical features
The 39 patients with ALK-positive histiocytosis comprised 24 females and 15 males and included 31 children and 8 adults (Table 1). Their age at presentation ranged from 0 days to 41 years: 1 infant had hepatosplenomegaly, anemia, and thrombocytopenia only 7 hours after birth.47 Moreover, the 31 pediatric cases consisted of 21 children of 3 years or younger, including 13 infants (age < 1 year). The 39 patients had diverse organ system manifestations (Figures 2-6) and were classified into 2 distinct clinical phenotypic groups: patients with multisystem disease (Group 1, n = 16) and patients with single-system disease (Group 2, n = 23). Within Group 1 were infants with systemic disease with liver and hematopoietic involvement as originally described29 (Group 1A, n = 6) and other patients with multisystemic disease, not fitting the original infantile disease presentation (Group 1B, n = 10).

Focal liver lesions in an infant with multisystemic disease with liver and hematopoietic involvement (Group 1A). (A) Ultrasound image showing 3 hypoechoic lesions in liver segment 3. (B-C) Coronal T2-weighted fat-suppressed MRI images showing multiple hyperintense lesions in the liver, including a large rounded lesion in liver segment 3 (C). (D) Coronal T2-weighted contrast-enhanced MRI image showing late contrast accumulation in the large rounded lesion in liver segment 3.
Focal liver lesions in an infant with multisystemic disease with liver and hematopoietic involvement (Group 1A). (A) Ultrasound image showing 3 hypoechoic lesions in liver segment 3. (B-C) Coronal T2-weighted fat-suppressed MRI images showing multiple hyperintense lesions in the liver, including a large rounded lesion in liver segment 3 (C). (D) Coronal T2-weighted contrast-enhanced MRI image showing late contrast accumulation in the large rounded lesion in liver segment 3.
Infants with systemic disease with liver and hematopoietic involvement (Group 1A)
Patients in Group 1A ranged from 0 days to 5 months of age and presented with hepatomegaly, anemia, and thrombocytopenia. In addition, 5/6 had splenomegaly, 5/6 displayed leukocytosis, and some had measurable liver dysfunction, such as elevated liver enzymes, high bilirubin, and/or decreased serum protein and albumin levels. In 3 cases, focal lesions were seen in the liver (Figure 2). Coagulation profiles were available for 4/6 patients (supplemental Table 2), revealing prolonged prothrombin time, activated partial thromboplastin time, and decreased serum fibrinogen in Case 3. Small numbers of ALK+ histiocytes were observed in the bone marrow of all 5 patients that had a bone marrow biopsy performed. Regarding additional sites of disease, 2/6 had renal involvement,31 1/6 had interstitial lung involvement requiring prolonged oxygen supplementation,47 and 1/6 had skin involvement (Table 3).
Other patients with multisystemic disease (Group 1B)
Group 1B contained 5 children and 5 adults who presented with disseminated disease involving various organs (Table 1), including 3 infants without hematopoietic involvement. The most commonly involved organs were the nervous system (7/10), bone (7/10), lungs (7/10), liver (5/10), skin (4/10), and lymph nodes (4/10). All 3 pediatric cases with neurologic involvement had multiple masses in the brain (Figure 4A-E); adult cases had lesions of cranial and/or spinal nerves (3/4; Figure 4G-I), intramedullary spinal cord and/or leptomeningeal enhancement (2/4; Figure 4F), and/or parenchymal brain masses (1/4). Reminiscent of the localization of bone lesions in ECD, bone involvement included bilateral lesions in femora and/or tibiae in 7/7 patients (Figure 3A-D). Lung involvement was uniformly characterized by pulmonary nodules, ranging in size from micronodules to significant tumors (Figure 3H-K). All 5 patients with liver involvement had focal lesions on imaging.
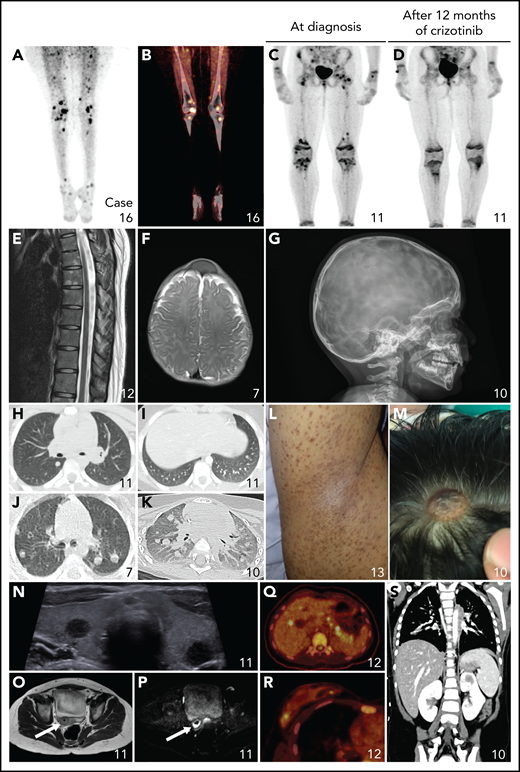
Nonneurologic disease manifestations in ALK-positive histiocytosis patients from Group 1B. (A-D) Fluorodeoxyglucose PET-CT images showing bilateral hypermetabolic long bone involvement, reminiscent of ECD, with objective metabolic response in Case 11 after 12 months of crizotinib. (E) Sagittal image of the contrast-enhanced MRI scan of the spine showing multiple hyperintense lesions in the vertebral bodies. (F-G) Axial MRI image (F) and lateral conventional radiograph (G) showing skull lesions in 2 children, with an appearance reminiscent of LCH. (H-K) Axial CT images showing nodular pulmonary involvement in 3 pediatric cases. (L) Photograph of the right axilla of an adult with a brown maculopapular exanthema that coalesces into plaques and predominates in the axillae and flanks, reminiscent of xanthoma disseminatum. (M) Photograph showing 1 of multiple scalp skin lesions in a child, which can also be observed on the MRI of the head (Figure 4A). (N) Ultrasound image demonstrating round, hypoechoic lesions in both lobes of the thyroid gland. (O-P) Axial T2-weighted (O) and diffusion-weighted (P) pelvic MRI images showing a cervical tumor with restricted diffusion in a child that presented with menorrhagia and irregular vaginal bleeding. (Q-R) Axial PET-CT images showing hypermetabolic focal lesions in the liver and pancreas (Q) and in the breast (R). (S) Coronal CT image showing a focal lesion in the left kidney.
Nonneurologic disease manifestations in ALK-positive histiocytosis patients from Group 1B. (A-D) Fluorodeoxyglucose PET-CT images showing bilateral hypermetabolic long bone involvement, reminiscent of ECD, with objective metabolic response in Case 11 after 12 months of crizotinib. (E) Sagittal image of the contrast-enhanced MRI scan of the spine showing multiple hyperintense lesions in the vertebral bodies. (F-G) Axial MRI image (F) and lateral conventional radiograph (G) showing skull lesions in 2 children, with an appearance reminiscent of LCH. (H-K) Axial CT images showing nodular pulmonary involvement in 3 pediatric cases. (L) Photograph of the right axilla of an adult with a brown maculopapular exanthema that coalesces into plaques and predominates in the axillae and flanks, reminiscent of xanthoma disseminatum. (M) Photograph showing 1 of multiple scalp skin lesions in a child, which can also be observed on the MRI of the head (Figure 4A). (N) Ultrasound image demonstrating round, hypoechoic lesions in both lobes of the thyroid gland. (O-P) Axial T2-weighted (O) and diffusion-weighted (P) pelvic MRI images showing a cervical tumor with restricted diffusion in a child that presented with menorrhagia and irregular vaginal bleeding. (Q-R) Axial PET-CT images showing hypermetabolic focal lesions in the liver and pancreas (Q) and in the breast (R). (S) Coronal CT image showing a focal lesion in the left kidney.

Neurologic involvement in ALK-positive histiocytosis patients from Group 1B or 2. (A-E) Axial images of the T1-weighted contrast-enhanced MRI scans of the heads of 2 pediatric cases with multiple solid brain tumors before and after treatment with ALK inhibition, demonstrating robust responses in both. (F) Sagittal image of the T1-weighted contrast-enhanced MRI scan of the spine showing leptomeningeal contrast enhancement along the descending cauda equina nerve roots. (G-I) Axial images of successive fluorodeoxyglucose PET-CT scans showing partial and complete response of a neuroforaminal tumor at level L5 after 2 cycles of cladribine (H) and subsequent treatment with alectinib (I), respectively. Coronal images (not shown) demonstrated that the tumor followed the course of the exiting nerves, highly reminiscent of nerve sheath tumors such as neurofibromas. (J-N) Axial images of successive T1-weighted contrast-enhanced MRI scans of the head of a child with a left insula tumor before and after subtotal resection and successful treatment with alectinib. (O-Q) Axial images of the T1-weighted contrast-enhanced MRI scans of the head of a child with a left oculomotor nerve tumor, demonstrating slight regression but continued contrast enhancement of the tumor after treatment with vinblastine/prednisone-based chemotherapy. (R) Coronal image of the T1-weighted contrast-enhanced MRI scan of the head showing a 30 × 25 × 34 mm large tumor with contrast enhancement in the prepontine cistern that followed the course of the trigeminal nerve and caused pressure on the pons. (S) Sagittal image of the T1-weighted contrast-enhanced MRI scan of the cervical spine showing a large (18 × 24 × 45 mm) intradural extramedullary tumor at level C1-C2.
Neurologic involvement in ALK-positive histiocytosis patients from Group 1B or 2. (A-E) Axial images of the T1-weighted contrast-enhanced MRI scans of the heads of 2 pediatric cases with multiple solid brain tumors before and after treatment with ALK inhibition, demonstrating robust responses in both. (F) Sagittal image of the T1-weighted contrast-enhanced MRI scan of the spine showing leptomeningeal contrast enhancement along the descending cauda equina nerve roots. (G-I) Axial images of successive fluorodeoxyglucose PET-CT scans showing partial and complete response of a neuroforaminal tumor at level L5 after 2 cycles of cladribine (H) and subsequent treatment with alectinib (I), respectively. Coronal images (not shown) demonstrated that the tumor followed the course of the exiting nerves, highly reminiscent of nerve sheath tumors such as neurofibromas. (J-N) Axial images of successive T1-weighted contrast-enhanced MRI scans of the head of a child with a left insula tumor before and after subtotal resection and successful treatment with alectinib. (O-Q) Axial images of the T1-weighted contrast-enhanced MRI scans of the head of a child with a left oculomotor nerve tumor, demonstrating slight regression but continued contrast enhancement of the tumor after treatment with vinblastine/prednisone-based chemotherapy. (R) Coronal image of the T1-weighted contrast-enhanced MRI scan of the head showing a 30 × 25 × 34 mm large tumor with contrast enhancement in the prepontine cistern that followed the course of the trigeminal nerve and caused pressure on the pons. (S) Sagittal image of the T1-weighted contrast-enhanced MRI scan of the cervical spine showing a large (18 × 24 × 45 mm) intradural extramedullary tumor at level C1-C2.
Patients with single-system disease (Group 2)
Patients in Group 2 were 20 children and 3 adults, including 12 cases with neurologic involvement (52%). Patients with neurologic involvement presented with diverse neurological symptoms, including seizures (3/12), ataxia (3/12), headaches (3/12), vomiting (2/12), hypotonicity (2/12), unclear paroxysmal neurologic symptoms (“spells”) (1/12), torticollis (1/12), trigeminal neuralgia (1/12), paresis (1/12), and diplopia (1/12). Tumoral lesions were widely distributed in the central and peripheral nervous system (Figure 4J-S; Table 3). In addition, 2 patients had cerebrospinal fluid monocytosis, including 1 case with a very low level of ALK rearrangements in the cerebrospinal fluid by FISH analysis. The 11 patients with nonneurologic disease comprised 1 case with a single lung tumor, 2 cases with solitary bone lesions, and 8 children with localized skin lesions or soft tissue tumors in varying locations (Figure 5; Table 3).

Nonneurologic disease manifestations in ALK-positive histiocytosis patients with single-system disease (Group 2). (A-D) Successive fluorodeoxyglucose PET-CT images of an adult female with a large right clavicular tumor at diagnosis (A; time, 0) and after treatment with radiotherapy (B; time, 5.5 months), 6 weeks of vinblastine/prednisone-based chemotherapy (C; time, 9.5 months), and 2 months of alectinib (D; time, 14 months). (E-I) T1-weighted contrast-enhanced (E), T2-weighted (F), diffusion-weighted (G), apparent diffusion coefficient (H), and plain T1-weighted (I) MRI images of the left lower leg of a child at diagnosis showing a single soft tissue tumor that infiltrates the musculature and shows contrast enhancement and restricted diffusion. (J-K) Photographs of the retroauricular scalp lesion of an infant, with a clear change in clinical appearance after 3 months (K).
Nonneurologic disease manifestations in ALK-positive histiocytosis patients with single-system disease (Group 2). (A-D) Successive fluorodeoxyglucose PET-CT images of an adult female with a large right clavicular tumor at diagnosis (A; time, 0) and after treatment with radiotherapy (B; time, 5.5 months), 6 weeks of vinblastine/prednisone-based chemotherapy (C; time, 9.5 months), and 2 months of alectinib (D; time, 14 months). (E-I) T1-weighted contrast-enhanced (E), T2-weighted (F), diffusion-weighted (G), apparent diffusion coefficient (H), and plain T1-weighted (I) MRI images of the left lower leg of a child at diagnosis showing a single soft tissue tumor that infiltrates the musculature and shows contrast enhancement and restricted diffusion. (J-K) Photographs of the retroauricular scalp lesion of an infant, with a clear change in clinical appearance after 3 months (K).
Treatments
Group 1A
Two of 6 patients did not receive histiocytosis-directed therapy but were managed with supportive care including transfusions (Table 2). In both cases, spontaneous resolution of disease was observed. Three other patients were treated with chemotherapy. Two experienced progressive disease, including 1 patient that had worsening of ascites and died of coagulopathy and sepsis. The other received second-line systemic therapy with dexamethasone, anakinra, and azathioprine and obtained a complete response. The third patient achieved a partial response to first-line chemotherapy, relapsed with biopsy-proven renal involvement at 5 months after completion of treatment, and subsequently received a second round of chemotherapy.31 The remaining patient was lost to follow-up.
Treatments and outcomes of the different clinical groups of ALK-positive histiocytosis and the overall cohort
| Group 1A (n = 6) | Group 1B (n = 10) | Group 2 (n = 23) | Overall (n = 39) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Number of cases | Objective response | Progression or relapse | Number of cases | Objective response | Progression or relapse | Number of cases | Objective response | Progression or relapse | Number of cases | Objective response | Progression or relapse | |
| First-line | ||||||||||||
| Conventional systemic therapy | 3 (50%) | 1/3 (33%) | 3/3 (100%) | 6 (60%) | 3/6 (50%) | 2/6 (33%) | 4 (17%)† | 3/4 (75%) | 1/4 (25%) | 13 (33%) | 7/13 (54%) | 6/13 (46%) |
| Surgical resection | 13 (57%)‡ | 11/13 (85%) | 2/13 (15%) | 13 (33%) | 11/13 (85%) | 2/13 (15%) | ||||||
| ALK inhibition | 4 (40%)* | 4/4 (100%) | 0/4 (0%) | 1 (4%) | 1/1 (100%) | 0/1 (0%) | 5 (13%) | 5/5 (100%) | 0/5 (0%) | |||
| Observation & supportive care | 2 (33%) | 2/2 (100%) | 0/2 (0%) | 1 (4%) | 1/1 (100%) | 0/1 (0%) | 3 (8%) | 3/3 (100%) | 0/3 (0%) | |||
| Radiotherapy | 1 (4%) | 1/1 (100%) | 0/1 (0%) | 1 (3%) | 1/1 (100%) | 0/1 (0%) | ||||||
| Unknown or not treated yet | 1 (17%) | 3 (13%) | 4 (10%) | |||||||||
| Second- and further-line | ||||||||||||
| ALK inhibition | 3 (30%) | 3/3 (100%) | 0/3 (0%) | 3 (13%) | 3/3 (100%) | 0/3 (0%) | 6 (15%) | 6/6 (100%) | 0/6 (0%) | |||
| Conventional systemic therapy | 3 (50%) | 2/3 (67%) | 1/3 (33%) | 2 (9%) | 0/2 (0%) | 2/2 (100%) | 5 (13%) | 2/5 (40%) | 3/5 (60%) | |||
| Group 1A (n = 6) | Group 1B (n = 10) | Group 2 (n = 23) | Overall (n = 39) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Number of cases | Objective response | Progression or relapse | Number of cases | Objective response | Progression or relapse | Number of cases | Objective response | Progression or relapse | Number of cases | Objective response | Progression or relapse | |
| First-line | ||||||||||||
| Conventional systemic therapy | 3 (50%) | 1/3 (33%) | 3/3 (100%) | 6 (60%) | 3/6 (50%) | 2/6 (33%) | 4 (17%)† | 3/4 (75%) | 1/4 (25%) | 13 (33%) | 7/13 (54%) | 6/13 (46%) |
| Surgical resection | 13 (57%)‡ | 11/13 (85%) | 2/13 (15%) | 13 (33%) | 11/13 (85%) | 2/13 (15%) | ||||||
| ALK inhibition | 4 (40%)* | 4/4 (100%) | 0/4 (0%) | 1 (4%) | 1/1 (100%) | 0/1 (0%) | 5 (13%) | 5/5 (100%) | 0/5 (0%) | |||
| Observation & supportive care | 2 (33%) | 2/2 (100%) | 0/2 (0%) | 1 (4%) | 1/1 (100%) | 0/1 (0%) | 3 (8%) | 3/3 (100%) | 0/3 (0%) | |||
| Radiotherapy | 1 (4%) | 1/1 (100%) | 0/1 (0%) | 1 (3%) | 1/1 (100%) | 0/1 (0%) | ||||||
| Unknown or not treated yet | 1 (17%) | 3 (13%) | 4 (10%) | |||||||||
| Second- and further-line | ||||||||||||
| ALK inhibition | 3 (30%) | 3/3 (100%) | 0/3 (0%) | 3 (13%) | 3/3 (100%) | 0/3 (0%) | 6 (15%) | 6/6 (100%) | 0/6 (0%) | |||
| Conventional systemic therapy | 3 (50%) | 2/3 (67%) | 1/3 (33%) | 2 (9%) | 0/2 (0%) | 2/2 (100%) | 5 (13%) | 2/5 (40%) | 3/5 (60%) | |||
Including 2 patients treated with a combination of ALK inhibition and chemotherapy.
Including 2 patients treated with chemotherapy following surgical resection.
Including 2 patients who received systemic corticosteroids before surgical resection.
Group 1B
First-line therapy consisted of conventional systemic therapy alone in 6/10 patients and ALK inhibition with or without conventional therapy in 4/10 patients (Table 2). Conventional systemic therapy led to an objective response in 3/6 patients, 2 of whom had sustained complete responses to chemotherapy and 1 of whom died of (likely immunosuppression-related) sepsis following a partial response to high-dose corticosteroids (Table 3). The 3/6 patients with stable or progressive disease following conventional systemic therapy were treated with ALK inhibition as second-line therapy, with objective responses in all 3. ALK inhibition, with or without chemotherapy, led to an objective response in 4/4 patients in the first-line setting. Responses to ALK inhibition were durable in 7/7 patients, with no events of progression or relapse on treatment (Figure 7).
Individual patient data of cases with ALK-positive histiocytosis (n = 39) or atypical ALK-rearranged histiocyte-rich tumors (n = 3)
| No. | Gene fusion | Sex | Age* | Sites of disease | First-line treatment | Response | Progression /relapse | Second- and further-line treatment | Last response | Therapy ongoing | Outcome (follow-up†) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Group 1A: infants with multistemic disease with liver and hematopoietic involvement | |||||||||||
| 1‡ | N/A | F | 0 d | Liver, hematopoietic system, spleen, lung, possibly kidney | IVIG (2 d), corticosteroids (tapered after diagnosis), and supportive care (transfusions) | CR | No | — | — | No | Alive with no disease (3.5 y) |
| 2‡ | ALK-FISH+ | F | 27 d | Liver, hematopoietic system, spleen, + kidney at relapse | Chemotherapy (VBL/PRED, 6 mo) | PR | Yes | Chemotherapy | CR | No | Alive with no disease (7 y)§ |
| 3 | KIF5B-ALK | M | 1 mo | Liver, hematopoietic system, spleen, kidney, skin | Chemotherapy (VBL/DEX/MTX, 3 wk) | PD | Yes | Chemotherapy (2-CDA, 1 wk) | PD | No | Died of coagulopathy and sepsis (1 mo) |
| 4 | N/A | F | 2 mo | Liver, hematopoietic system, spleen | Supportive care (transfusions) | CR | No | — | — | No | Alive with no disease (4.5 y) |
| 5 | ALK-FISH+ | F | 4 mo | Liver, hematopoietic system | N/A | N/A | N/A | N/A | N/A | N/A | N/A (lost to follow-up) |
| 6 | CLTC-ALK | M | 5 mo | Liver, hematopoietic system, spleen | Corticosteroids (PRED 40mg/m2, 2.5 wk), followed by chemotherapy (VBL + tapered PRED, 2.5 wk) | PD | Yes | Dexamethasone + anakinra (1 mo), followed by anakinra (3 mo) + azathioprine (4.5 mo) | CR | No | Alive with no disease (4 y) |
| Group 1B: other patients with multisystemic disease | |||||||||||
| 7 | TPM3-ALK | F | 3 mo | Bone, lung, liver | Chemotherapy (VBL/PRED/6MP, 18 mo) | CR | No | — | — | No | Alive with no disease (2 y) |
| 8 | ALK-FISH+ | F | 9 mo | Lung, skin, kidney | Chemotherapy (VBL/PRED, 12 mo) | CR | No | — | — | No | Alive with no disease (2 y) |
| 9‡ | KIF5B-ALK | M | 10 mo | CNS, lung, liver, soft tissue (peritoneum) | Chemotherapy (VBL/PRED), combined with ALK inhibition (alectinib) | CR | No | — | — | Yes (VBL/PRED + alectinib) | Alive with no disease (13 mo) |
| 10 | KIF5B-ALK | F | 2 y | CNS, bone, lung, liver, skin, soft tissue (perineal mass), lymph node, kidney, breast, pancreas | Chemotherapy (VBL/PRED, 6 wk induction) | SD | No | ALK inhibition (lorlatinib) | PR | Yes (lorlatinib) | Alive with regressive disease (21 mo) |
| 11 | KIF5B-ALK | F | 10 y | CNS, bone, lung, lymph node, cervix, thyroid, submandibular salivary gland | ALK inhibition (crizotinib) | CR | No | — | — | Yes (crizotinib) | Alive with no disease (2 y) |
| 12 | KIF5B-ALK | F | 19 y | CNS/PNS, bone, lung, liver, lymph node, breast, pancreas | Chemotherapy (2-CDA, 3 cycles), followed by ALK inhibition (alectinib) | PR | No | — | — | Yes (alectinib) | Alive with stable (PETnegative) bone lesions on MRI; other lesions regressed (2 y) |
| 13 | TFG-ALK | M | 21 y | Liver, skin, colorectum | Corticosteroids (high-dose) | PR | Yes‖ | — | — | No | Died of sepsis (2 mo) |
| 14 | KIF5B-ALK | F | 28 y | CNS/PNS, bone | ALK inhibition (alectinib) | PR | No | — | — | Yes (alectinib) | Alive with regressive (PETnegative) disease (9 mo) |
| 15 | KIF5B-ALK | M | 29 y | PNS (incl. cerebrospinal fluid pleocytosis), bone | Corticosteroids, followed by pegylated interferon-α (with escalation to 180μg/wk, 4 mo) | SD | No | ALK inhibition (brigatinib) | CR | Yes (brigatinib) | Alive with no disease (2.5 y) |
| 16‡ | KIF5B-ALK | F | 41 y | CNS, bone, lung, skin, soft tissue (omentum/peritoneum), lymph node | Interferon-α (16 mo) | PD | Yes | ALK inhibition (crizotinib) | PR | Yes (crizotinib) | Alive with regressive disease (6 y) |
| Group 2: single-system neurologic disease | |||||||||||
| 17 | KIF5B-ALK | F | 7 mo | CNS: thalamus | Corticosteroids, followed by ALK inhibition (lorlatinib) | PR | No | — | — | Yes (lorlatinib) | Alive with regressive disease (5 mo) |
| 18 | ALK-FISH+ | F | 9 mo | CNS: cerebellum | N/A | N/A | N/A | N/A | N/A | N/A | N/A (lost to follow-up) |
| 19 | KIF5B-ALK | M | 2.5 y | CNS: right medulla tumor extending through the foramen of Luschka | Chemotherapy (Headstart protocol, 1 cycle) | PD | Yes | ALK inhibition (alectinib) | CR | Yes (alectinib) | Alive with no disease (16 mo) |
| 20 | KIF5B-ALK | F | 3 y | CNS: cerebellum, cranial nerves, spinal cord, cerebrospinal fluid | Corticosteroids, followed by surgery (subtotal resection cerebellar tumor) | CR | No | — | — | No | Alive with no disease (12 mo) |
| 21 | KIF5B-ALK | F | 3 y | PNS: left oculomotor nerve with extraneural extension | Corticosteroids, followed by surgery (subtotal resection) and chemotherapy (VBL/PRED, 6 mo) | PR | No | — | — | No | Alive with mildly regressive disease (2.5 y) |
| 22 | KIF5B-ALK | F | 7 y | CNS: left medulla | Chemotherapy (Clofarabine, 6 cycles) | CR | No | — | — | No | Alive with no disease (6 mo) |
| 23‡ | KIF5B-ALK | F | 11 y | CNS: right frontal lobe | Surgery (total resection) | CR | No | — | — | No | Alive with no disease (9 mo) |
| 24 | ALK-FISH+ | M | 11 y | PNS: left trigeminal nerve | Surgery (subtotal resection) | CR | No | — | — | No | Alive with no disease (18 y) |
| 25 | KIF5B-ALK | M | 12 y | PNS: intradural extramedullary tumor at level C1-C2 | Surgery (total resection), followed by chemotherapy (VBL/PRED, 12 mo) | CR | No | — | — | No | Alive with no disease (2.5 y) |
| 26 | KIF5B-ALK | F | 13 y | CNS/PNS: left insula, cranial & spinal nerves, pituitary stalk, + cerebrospinal fluid at relapse | Surgery (subtotal resection insula tumor) | PD | Yes | ALK inhibition (alectinib) | PR | Yes (alectinib) | Alive with minor, regressive disease (2.5 y) |
| 27 | KIF5B-ALK | M | 20 y | PNS: 3 intradural extramedullary lesions at levels L3 and S1 | Not treated yet | Not yet evaluable | Not yet evaluable | — | — | No | Alive with active disease (7 mo) |
| 28 | KIF5B-ALK | M | 20 y | CNS: right frontal lobe | Surgery (total resection) | CR | No | — | — | No | Alive with no disease (10 mo) |
| Group 2: single-system nonneurologic disease | |||||||||||
| 29 | KIF5B-ALK | F | 6 mo | Skin: retroauricular scalp lesion | Active monitoring (4 mo), followed by surgery (subtotal resection) | CR | No | — | — | No | Alive with no disease (2 y) |
| 30 | KIF5B-ALK | M | 7 mo | Skin: papular lesion on the back | Surgery (total resection) | CR | No | — | — | No | Alive with no disease (13 mo) |
| 31 | KIF5B-ALK | F | 21 mo | Skin: midline posterior scalp lesion | Surgery (total resection) | CR | No | — | — | No | Alive with no disease (2 y) |
| 32 | KIF5B-ALK | M | 2 y | Soft tissue: subcutaneous tumor left lower leg with infiltration in the musculature | Active monitoring | PR | No | — | — | No | Alive with regressive disease (14 mo) |
| 33 | KIF5B-ALK | F | 3 y | Soft tissue: perineal mass | Surgery (total resection) | CR | No | — | — | No | Alive with no disease (14 mo) |
| 34‡ | KIF5B-ALK | M | 3 y | Soft tissue: subglottic mass | Corticosteroids, followed by surgery (total resection) | CR | No | — | — | No | Alive with no disease (3 y) |
| 35 | KIF5B-ALK | F | 10 y | Skin: single nodule on the right breast (7mm; exclusively dermal/cutaneous) | Surgery (total resection) | CR | No | — | — | No | Alive with no disease (1 mo) |
| 36 | KIF5B-ALK | F | 10 y | Bone: right scapular lesion with soft tissue extension | Surgery (total resection) | CR | No | — | — | No | Alive with no disease (5 y) |
| 37 | KIF5B-ALK | M | 11 y | Soft tissue: 2 tumors in omentum and mesenterium | Surgery (subtotal resection) | PD | Yes | Chemotherapy (VBL/PRED, 12 wk, IC-1/IC-2, → PD), followed by active monitoring¶ | SD | No | Alive with stable disease (15 mo) |
| 38 | EML4-ALK | M | 17 y | Lung: single mass (10 × 9 × 8 cm) within the left lower lobe extending into the pulmonary vein and left atrium | N/A | N/A | N/A | N/A | N/A | N/A | N/A (lost to follow-up) |
| 39 | DCTN1-ALK | F | 41 y | Bone: right clavicular lesion with significant soft tissue extension (total size >10 cm) | Radiotherapy (22 Gy in 12 fractions) | PR | No | ALK inhibition (crizotinib → severe anaphylaxis), followed by chemotherapy (VBL/PRED, 4 courses, → PD), and then ALK inhibition (alectinib) | PR | Yes (alectinib) | Alive with regressive disease (2 y) |
| Atypical: ALK-rearranged histiocyte-rich tumors | |||||||||||
| A1 | EML4-ALK | F | 51 y | CNS, bone, lung, + subcutaneous tumors at relapse | Surgery (L1 tumor), chemotherapy (TC, 1 cycle) and radiotherapy (32 Gy L1 tumor; 20 Gy whole brain) | PD | Yes | ALK inhibition (alectinib for 16 mo, followed by lorlatinib for 14 mo, and then ceritinib for 7 wk), followed by antalgic radiotherapy of 2 metastases and chemotherapy (VBL/PRED) | SD | Yes (VBL/Pred) | Alive with stable disease (3 y) |
| A2 | SQSTM1-ALK | F | 53 y | Soft tissue: large mesenteric tumor, + subcutaneous and liver tumors at relapse | Surgery (total resection) | PD | Yes | ALK inhibition (crizotinib) | SD | Yes (crizotinib) | Alive with stable disease (13 mo) |
| A3 | EML4-ALK | F | 4 y | Soft tissue: obstructing yellow nodule in the left main bronchus | Surgery (endoscopic resection) | N/A | Yes | Surgery (endoscopic reresection 1 and 2 y after first surgery) | CR | No | Alive with no disease (17 y) |
| No. | Gene fusion | Sex | Age* | Sites of disease | First-line treatment | Response | Progression /relapse | Second- and further-line treatment | Last response | Therapy ongoing | Outcome (follow-up†) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Group 1A: infants with multistemic disease with liver and hematopoietic involvement | |||||||||||
| 1‡ | N/A | F | 0 d | Liver, hematopoietic system, spleen, lung, possibly kidney | IVIG (2 d), corticosteroids (tapered after diagnosis), and supportive care (transfusions) | CR | No | — | — | No | Alive with no disease (3.5 y) |
| 2‡ | ALK-FISH+ | F | 27 d | Liver, hematopoietic system, spleen, + kidney at relapse | Chemotherapy (VBL/PRED, 6 mo) | PR | Yes | Chemotherapy | CR | No | Alive with no disease (7 y)§ |
| 3 | KIF5B-ALK | M | 1 mo | Liver, hematopoietic system, spleen, kidney, skin | Chemotherapy (VBL/DEX/MTX, 3 wk) | PD | Yes | Chemotherapy (2-CDA, 1 wk) | PD | No | Died of coagulopathy and sepsis (1 mo) |
| 4 | N/A | F | 2 mo | Liver, hematopoietic system, spleen | Supportive care (transfusions) | CR | No | — | — | No | Alive with no disease (4.5 y) |
| 5 | ALK-FISH+ | F | 4 mo | Liver, hematopoietic system | N/A | N/A | N/A | N/A | N/A | N/A | N/A (lost to follow-up) |
| 6 | CLTC-ALK | M | 5 mo | Liver, hematopoietic system, spleen | Corticosteroids (PRED 40mg/m2, 2.5 wk), followed by chemotherapy (VBL + tapered PRED, 2.5 wk) | PD | Yes | Dexamethasone + anakinra (1 mo), followed by anakinra (3 mo) + azathioprine (4.5 mo) | CR | No | Alive with no disease (4 y) |
| Group 1B: other patients with multisystemic disease | |||||||||||
| 7 | TPM3-ALK | F | 3 mo | Bone, lung, liver | Chemotherapy (VBL/PRED/6MP, 18 mo) | CR | No | — | — | No | Alive with no disease (2 y) |
| 8 | ALK-FISH+ | F | 9 mo | Lung, skin, kidney | Chemotherapy (VBL/PRED, 12 mo) | CR | No | — | — | No | Alive with no disease (2 y) |
| 9‡ | KIF5B-ALK | M | 10 mo | CNS, lung, liver, soft tissue (peritoneum) | Chemotherapy (VBL/PRED), combined with ALK inhibition (alectinib) | CR | No | — | — | Yes (VBL/PRED + alectinib) | Alive with no disease (13 mo) |
| 10 | KIF5B-ALK | F | 2 y | CNS, bone, lung, liver, skin, soft tissue (perineal mass), lymph node, kidney, breast, pancreas | Chemotherapy (VBL/PRED, 6 wk induction) | SD | No | ALK inhibition (lorlatinib) | PR | Yes (lorlatinib) | Alive with regressive disease (21 mo) |
| 11 | KIF5B-ALK | F | 10 y | CNS, bone, lung, lymph node, cervix, thyroid, submandibular salivary gland | ALK inhibition (crizotinib) | CR | No | — | — | Yes (crizotinib) | Alive with no disease (2 y) |
| 12 | KIF5B-ALK | F | 19 y | CNS/PNS, bone, lung, liver, lymph node, breast, pancreas | Chemotherapy (2-CDA, 3 cycles), followed by ALK inhibition (alectinib) | PR | No | — | — | Yes (alectinib) | Alive with stable (PETnegative) bone lesions on MRI; other lesions regressed (2 y) |
| 13 | TFG-ALK | M | 21 y | Liver, skin, colorectum | Corticosteroids (high-dose) | PR | Yes‖ | — | — | No | Died of sepsis (2 mo) |
| 14 | KIF5B-ALK | F | 28 y | CNS/PNS, bone | ALK inhibition (alectinib) | PR | No | — | — | Yes (alectinib) | Alive with regressive (PETnegative) disease (9 mo) |
| 15 | KIF5B-ALK | M | 29 y | PNS (incl. cerebrospinal fluid pleocytosis), bone | Corticosteroids, followed by pegylated interferon-α (with escalation to 180μg/wk, 4 mo) | SD | No | ALK inhibition (brigatinib) | CR | Yes (brigatinib) | Alive with no disease (2.5 y) |
| 16‡ | KIF5B-ALK | F | 41 y | CNS, bone, lung, skin, soft tissue (omentum/peritoneum), lymph node | Interferon-α (16 mo) | PD | Yes | ALK inhibition (crizotinib) | PR | Yes (crizotinib) | Alive with regressive disease (6 y) |
| Group 2: single-system neurologic disease | |||||||||||
| 17 | KIF5B-ALK | F | 7 mo | CNS: thalamus | Corticosteroids, followed by ALK inhibition (lorlatinib) | PR | No | — | — | Yes (lorlatinib) | Alive with regressive disease (5 mo) |
| 18 | ALK-FISH+ | F | 9 mo | CNS: cerebellum | N/A | N/A | N/A | N/A | N/A | N/A | N/A (lost to follow-up) |
| 19 | KIF5B-ALK | M | 2.5 y | CNS: right medulla tumor extending through the foramen of Luschka | Chemotherapy (Headstart protocol, 1 cycle) | PD | Yes | ALK inhibition (alectinib) | CR | Yes (alectinib) | Alive with no disease (16 mo) |
| 20 | KIF5B-ALK | F | 3 y | CNS: cerebellum, cranial nerves, spinal cord, cerebrospinal fluid | Corticosteroids, followed by surgery (subtotal resection cerebellar tumor) | CR | No | — | — | No | Alive with no disease (12 mo) |
| 21 | KIF5B-ALK | F | 3 y | PNS: left oculomotor nerve with extraneural extension | Corticosteroids, followed by surgery (subtotal resection) and chemotherapy (VBL/PRED, 6 mo) | PR | No | — | — | No | Alive with mildly regressive disease (2.5 y) |
| 22 | KIF5B-ALK | F | 7 y | CNS: left medulla | Chemotherapy (Clofarabine, 6 cycles) | CR | No | — | — | No | Alive with no disease (6 mo) |
| 23‡ | KIF5B-ALK | F | 11 y | CNS: right frontal lobe | Surgery (total resection) | CR | No | — | — | No | Alive with no disease (9 mo) |
| 24 | ALK-FISH+ | M | 11 y | PNS: left trigeminal nerve | Surgery (subtotal resection) | CR | No | — | — | No | Alive with no disease (18 y) |
| 25 | KIF5B-ALK | M | 12 y | PNS: intradural extramedullary tumor at level C1-C2 | Surgery (total resection), followed by chemotherapy (VBL/PRED, 12 mo) | CR | No | — | — | No | Alive with no disease (2.5 y) |
| 26 | KIF5B-ALK | F | 13 y | CNS/PNS: left insula, cranial & spinal nerves, pituitary stalk, + cerebrospinal fluid at relapse | Surgery (subtotal resection insula tumor) | PD | Yes | ALK inhibition (alectinib) | PR | Yes (alectinib) | Alive with minor, regressive disease (2.5 y) |
| 27 | KIF5B-ALK | M | 20 y | PNS: 3 intradural extramedullary lesions at levels L3 and S1 | Not treated yet | Not yet evaluable | Not yet evaluable | — | — | No | Alive with active disease (7 mo) |
| 28 | KIF5B-ALK | M | 20 y | CNS: right frontal lobe | Surgery (total resection) | CR | No | — | — | No | Alive with no disease (10 mo) |
| Group 2: single-system nonneurologic disease | |||||||||||
| 29 | KIF5B-ALK | F | 6 mo | Skin: retroauricular scalp lesion | Active monitoring (4 mo), followed by surgery (subtotal resection) | CR | No | — | — | No | Alive with no disease (2 y) |
| 30 | KIF5B-ALK | M | 7 mo | Skin: papular lesion on the back | Surgery (total resection) | CR | No | — | — | No | Alive with no disease (13 mo) |
| 31 | KIF5B-ALK | F | 21 mo | Skin: midline posterior scalp lesion | Surgery (total resection) | CR | No | — | — | No | Alive with no disease (2 y) |
| 32 | KIF5B-ALK | M | 2 y | Soft tissue: subcutaneous tumor left lower leg with infiltration in the musculature | Active monitoring | PR | No | — | — | No | Alive with regressive disease (14 mo) |
| 33 | KIF5B-ALK | F | 3 y | Soft tissue: perineal mass | Surgery (total resection) | CR | No | — | — | No | Alive with no disease (14 mo) |
| 34‡ | KIF5B-ALK | M | 3 y | Soft tissue: subglottic mass | Corticosteroids, followed by surgery (total resection) | CR | No | — | — | No | Alive with no disease (3 y) |
| 35 | KIF5B-ALK | F | 10 y | Skin: single nodule on the right breast (7mm; exclusively dermal/cutaneous) | Surgery (total resection) | CR | No | — | — | No | Alive with no disease (1 mo) |
| 36 | KIF5B-ALK | F | 10 y | Bone: right scapular lesion with soft tissue extension | Surgery (total resection) | CR | No | — | — | No | Alive with no disease (5 y) |
| 37 | KIF5B-ALK | M | 11 y | Soft tissue: 2 tumors in omentum and mesenterium | Surgery (subtotal resection) | PD | Yes | Chemotherapy (VBL/PRED, 12 wk, IC-1/IC-2, → PD), followed by active monitoring¶ | SD | No | Alive with stable disease (15 mo) |
| 38 | EML4-ALK | M | 17 y | Lung: single mass (10 × 9 × 8 cm) within the left lower lobe extending into the pulmonary vein and left atrium | N/A | N/A | N/A | N/A | N/A | N/A | N/A (lost to follow-up) |
| 39 | DCTN1-ALK | F | 41 y | Bone: right clavicular lesion with significant soft tissue extension (total size >10 cm) | Radiotherapy (22 Gy in 12 fractions) | PR | No | ALK inhibition (crizotinib → severe anaphylaxis), followed by chemotherapy (VBL/PRED, 4 courses, → PD), and then ALK inhibition (alectinib) | PR | Yes (alectinib) | Alive with regressive disease (2 y) |
| Atypical: ALK-rearranged histiocyte-rich tumors | |||||||||||
| A1 | EML4-ALK | F | 51 y | CNS, bone, lung, + subcutaneous tumors at relapse | Surgery (L1 tumor), chemotherapy (TC, 1 cycle) and radiotherapy (32 Gy L1 tumor; 20 Gy whole brain) | PD | Yes | ALK inhibition (alectinib for 16 mo, followed by lorlatinib for 14 mo, and then ceritinib for 7 wk), followed by antalgic radiotherapy of 2 metastases and chemotherapy (VBL/PRED) | SD | Yes (VBL/Pred) | Alive with stable disease (3 y) |
| A2 | SQSTM1-ALK | F | 53 y | Soft tissue: large mesenteric tumor, + subcutaneous and liver tumors at relapse | Surgery (total resection) | PD | Yes | ALK inhibition (crizotinib) | SD | Yes (crizotinib) | Alive with stable disease (13 mo) |
| A3 | EML4-ALK | F | 4 y | Soft tissue: obstructing yellow nodule in the left main bronchus | Surgery (endoscopic resection) | N/A | Yes | Surgery (endoscopic reresection 1 and 2 y after first surgery) | CR | No | Alive with no disease (17 y) |
2-CDA, cladribine; 6MP, mercaptopurine; CNS, central nervous system; CR, complete response; F, female; Gy, gray; IVIG, intravenous immunoglobulin; M, male; N/A, not available; PD, progressive disease; PNS, peripheral nervous system; PR, partial response; PRED, prednisone; SD, stable disease; TC, paclitaxel and cyclophosphamide; VBL, vinblastine.
Age at presentation in hospital.
Follow-up since presentation in hospital.
This patient experiences chronic renal insufficiency, hypertension, and hypergammaglobulinemia.
Death.
Abdominal MRI after chemotherapy demonstrated disease progression. Subsequent abdominal ultrasound exams at 2 and 4 mo after abdominal MRI showed stable disease; the patient has been asymptomatic since he received chemotherapy. Therefore, no second-line therapy was given at last follow-up, but active monitoring was executed. Active monitoring was performed using abdominal ultrasound because of significant distress of the child during MRI.

Body diagram showing recurrent anatomic sites of involvement of ALK-positive histiocytosis.
Body diagram showing recurrent anatomic sites of involvement of ALK-positive histiocytosis.

Swimmer plot of outcomes in patients with ALK-positive histiocytosis (n = 11) or atypical ALK-rearranged histiocyte-rich tumors (n = 2) treated with ALK inhibition. ALK inhibition was initiated at timepoint zero. Median time on ALK inhibition was 16 months in ALK-positive histiocytosis patients (range 3-43 months). Responses were measured by CT, MRI, and/or PET-CT in all patients. Dose reductions were 67% (90 mg brigatinib/d → 30 mg/d) and 50% (1200 mg alectinib/d → 900 mg/d → 600 mg/d) in Case 15 and Case 26, respectively. Case 39 developed a severe (grade 3) anaphylactic shock on the first day of crizotinib administration, requiring the patient to be resuscitated. The patient subsequently received vinblastine/prednisone-based chemotherapy with progressive disease and then switched to alectinib with objective response after 2 months. Case A1 developed a subcutaneous gluteal metastasis during treatment with alectinib (supplemental Figure 3C), which was found to harbor an ALK p.I1171N mutation, a mutation known to confer secondary resistance to alectinib.78,79 Therefore, the patient switched to lorlatinib and later to ceritinib after repeated progressive disease. Due to continuing progressive disease during treatment with ceritinib, the patient recently stopped ceritinib, received 3 weeks of bridging therapy with lorlatinib during antalgic radiotherapy of 2 metastases, and subsequently started vinblastine/prednisone-based chemotherapy. VBL/PRED, vinblastine and prednisone-based chemotherapy.
Swimmer plot of outcomes in patients with ALK-positive histiocytosis (n = 11) or atypical ALK-rearranged histiocyte-rich tumors (n = 2) treated with ALK inhibition. ALK inhibition was initiated at timepoint zero. Median time on ALK inhibition was 16 months in ALK-positive histiocytosis patients (range 3-43 months). Responses were measured by CT, MRI, and/or PET-CT in all patients. Dose reductions were 67% (90 mg brigatinib/d → 30 mg/d) and 50% (1200 mg alectinib/d → 900 mg/d → 600 mg/d) in Case 15 and Case 26, respectively. Case 39 developed a severe (grade 3) anaphylactic shock on the first day of crizotinib administration, requiring the patient to be resuscitated. The patient subsequently received vinblastine/prednisone-based chemotherapy with progressive disease and then switched to alectinib with objective response after 2 months. Case A1 developed a subcutaneous gluteal metastasis during treatment with alectinib (supplemental Figure 3C), which was found to harbor an ALK p.I1171N mutation, a mutation known to confer secondary resistance to alectinib.78,79 Therefore, the patient switched to lorlatinib and later to ceritinib after repeated progressive disease. Due to continuing progressive disease during treatment with ceritinib, the patient recently stopped ceritinib, received 3 weeks of bridging therapy with lorlatinib during antalgic radiotherapy of 2 metastases, and subsequently started vinblastine/prednisone-based chemotherapy. VBL/PRED, vinblastine and prednisone-based chemotherapy.
Group 2
First-line treatment consisted of surgical resection in 13/23, radiation in 1/23, conventional systemic therapy with or without local therapy in 4/23, and ALK inhibition in 1/23 patients (Table 2). Furthermore, 1/23 patients was observed without treatment, 1/23 was not treated yet, and 2/23 were lost to follow-up. Of those with local therapy, 2/14 had progressive disease. Of patients treated with conventional systemic therapy, 3/4 had an objective response, and 1/4 had progressive disease. The patient treated with ALK inhibition had an objective response. Second-line treatment was given to 4 patients. ALK inhibition was administered in 2/4 patients with objective response, whereas the 2 others received chemotherapy with progressive disease. Third-line treatment with ALK inhibition was initiated in 1 of these patients with objective response (Figure 5A-D). Responses to ALK inhibition were durable in 4/4 patients (Figure 7).
Histopathologic features
The morphology of ALK-positive histiocytoses varied and included classic xanthogranuloma features with plump foamy histiocytes and variable Touton giant cells in 12/39 (31%) patients (Figure 8A-B) to a more densely cellular, monomorphic appearance without lipidized histiocytes in the majority of cases (Figure 8C,E), some with more spindled/streaming or epithelioid histiocyte morphology (supplemental Table 1). Nuclear features included ovoid nuclei that frequently displayed either a slight fold or indentation (ie, “cup shape”) of its nuclear contour. Mild atypia was limited to 6/39 cases, often with an epithelioid phenotype, which can be confused with malignant histiocytosis; however, mitotic counts and Ki-67 proliferation indexes were low to moderate. Bone marrow involvement was typically a small amount and not a diffuse infiltrative process as in leukemia. The morphology was that of plump histiocytes with indented nuclei and a juvenile xanthogranuloma (JXG)-like phenotype in normo- to hypercellular bone marrow with relatively preserved hematopoiesis and occasional eosinophilia, mild myelofibrosis, and/or megakaryocytic hyperplasia.

Histopathologic features of ALK-positive histiocytosis. (A) Photomicrograph of the hematoxylin and eosin (HE)-stained slide of a frontal bone tumor (Case 7; original magnification ×200) with classic xanthogranuloma morphology including many Touton giant cells. (B) HE image of a spinal nerve root tumor (Case 15; original magnification ×200) showing abundant lipidized (“foamy”) histiocytes. (C) HE image (Case 31; original magnification ×400) showing a more monomorphic histiocytic infiltrate in the skin dissecting through the dermal collagen bundles. (D) HE image of a liver biopsy (Case 4; original magnification ×400) showing sinusoidal infiltration by large histiocytes (indicated by black arrows) with ALK immunoreactivity (inlet). (E) HE image of a CNS tumor (Case 18; original magnification ×400) with a monomorphic, dense infiltrate of histiocytes that demonstrate separated red and green signals on ALK break-apart FISH analysis (inlet). (F) HE image of a CNS lesion (Case 20; ×100) showing marked infiltration of the perivascular (“Virchow-Robin”) spaces by histiocytes with clear CD163 immunoreactivity (inlet). (G) CD163 immunostain of a CNS tumor (Case 18; original magnification ×200) showing diffuse strong expression by the monomorphic histiocytic infiltrate. (H) ALK immunostain (Case 7; original magnification ×200) showing strong cytoplasmic and membranous staining of lesional histiocytes and Touton giant cells. (I) ALK immunostain of a breast tumor (Case 12; original magnification ×400) showing focal, exclusive dot-like immunoreactivity that could be misinterpreted as negative. (J) S100 immunostain of a liver biopsy (Case 4; ×200) showing immunoreactivity by the large sinusoidal histiocytes. (K) P-ERK immunostain (Case 15; original magnification ×400) showing diffuse positive staining by the lesional cells, as well as clear emperipolesis (intact intracytoplasmic leukocytes). (L) Cyclin D1 immunostain of an oculomotor nerve tumor (Case 21; original magnification ×200) showing cytoplasmic and strong nuclear staining in histiocytes with frequent nuclear indentations.
Histopathologic features of ALK-positive histiocytosis. (A) Photomicrograph of the hematoxylin and eosin (HE)-stained slide of a frontal bone tumor (Case 7; original magnification ×200) with classic xanthogranuloma morphology including many Touton giant cells. (B) HE image of a spinal nerve root tumor (Case 15; original magnification ×200) showing abundant lipidized (“foamy”) histiocytes. (C) HE image (Case 31; original magnification ×400) showing a more monomorphic histiocytic infiltrate in the skin dissecting through the dermal collagen bundles. (D) HE image of a liver biopsy (Case 4; original magnification ×400) showing sinusoidal infiltration by large histiocytes (indicated by black arrows) with ALK immunoreactivity (inlet). (E) HE image of a CNS tumor (Case 18; original magnification ×400) with a monomorphic, dense infiltrate of histiocytes that demonstrate separated red and green signals on ALK break-apart FISH analysis (inlet). (F) HE image of a CNS lesion (Case 20; ×100) showing marked infiltration of the perivascular (“Virchow-Robin”) spaces by histiocytes with clear CD163 immunoreactivity (inlet). (G) CD163 immunostain of a CNS tumor (Case 18; original magnification ×200) showing diffuse strong expression by the monomorphic histiocytic infiltrate. (H) ALK immunostain (Case 7; original magnification ×200) showing strong cytoplasmic and membranous staining of lesional histiocytes and Touton giant cells. (I) ALK immunostain of a breast tumor (Case 12; original magnification ×400) showing focal, exclusive dot-like immunoreactivity that could be misinterpreted as negative. (J) S100 immunostain of a liver biopsy (Case 4; ×200) showing immunoreactivity by the large sinusoidal histiocytes. (K) P-ERK immunostain (Case 15; original magnification ×400) showing diffuse positive staining by the lesional cells, as well as clear emperipolesis (intact intracytoplasmic leukocytes). (L) Cyclin D1 immunostain of an oculomotor nerve tumor (Case 21; original magnification ×200) showing cytoplasmic and strong nuclear staining in histiocytes with frequent nuclear indentations.
The ALK staining pattern did not appear to correlate with molecular alteration as the frequent KIF5B-ALK fusion was noted with all stain patterns; however, no (convincing) nuclear ALK staining was observed. Four cases displayed focal weak ALK staining and 3/39 had exclusive cytoplasmic Golgi dot-like staining in the lesional histiocytes (Figure 8I; Table 4) despite confirmed ALK fusions. Almost half of cases had variable Rosai-Dorfman disease–like histologic features including S100 expression (18/39) and emperipolesis (13/39). When tested for OCT-2 expression,58 14/23 (61%) were positive. When tested for p-ERK (18/39) and Cyclin D1 (19/39) expression (supplemental Table 1), the lesional histiocytic cells often showed corresponding, strong expression (Figure 8K-L).
Histopathologic and molecular characteristics of ALK-positive histiocytoses
| No. | ALK rearrangement | Analysis method(s) | Classic XG-like | Emperipolesis | ALK IHC | |
|---|---|---|---|---|---|---|
| Staining | Staining pattern | |||||
| Group 1A | ||||||
| 1 | Not confirmed | ALK-FISH negative; KIF5B-ALK RT-PCR negative; no RNA-seq (insufficient material) | No | Yes | Positive | Light cytoplasmic; membranous |
| 2 | ALK-FISH+ | FISH | No | Yes abundant | Positive | Diffuse cytoplasmic; membranous |
| 3 | KIF5B-ALK | Archer FusionPlex targeted RNA-seq | No | No | Positive | Granular cytoplasmic with focal Golgi dot-like accentuation; no membranous |
| 4 | Not confirmed | ALK-FISH negative; no RNA-seq (insufficient material) | Yes | Yes abundant | Positive | Scant cytoplasmic; strong membranous |
| 5 | ALK-FISH+ | FISH | Yes | Yes | Positive | Weak cytoplasmic; strong membranous |
| 6 | CLTC-ALK | Archer FusionPlex targeted RNA-seq | No | Yes | Positive | Diffuse dark cytoplasmic (clone 5A4) to more light granular cytoplasmic (clone ALK1); membranous (both) |
| Group 1B | ||||||
| 7 | TPM3-ALK | Archer FusionPlex targeted RNA-seq | Yes | Yes | Positive | Diffuse cytoplasmic with Golgi dot-like accentuation; membranous |
| 8 | ALK-FISH+ | FISH | No | No | Positive | Diffuse granular cytoplasmic with rare Golgi dot-like accentuation; no membranous |
| 9 | KIF5B-ALK | Illumina Childhood Cancer targeted RNA-seq | No | No | Weak | Very weak diffuse cytoplasmic; no membranous |
| 10 | KIF5B-ALK | Illumina Trusight targeted RNA-seq | No | No | Positive | Diffuse dark cytoplasmic (clone D5F3) to more light, granular cytoplasmic with Golgi dot-like accentuation (clone ALK1); no membranous |
| 11 | KIF5B-ALK | Archer FusionPlex targeted RNA-seq | No | No | Positive | Diffuse cytoplasmic; no membranous |
| 12 | KIF5B-ALK | KIF5B-ALK RT-PCT positive | No | No | Exclusive dot-like | Golgi dot-like cytoplasmic only; no membranous |
| 13 | TFG-ALK | Archer FusionPlex targeted RNA-seq | Yes | No | Focal, weak | Very weak cytoplasmic blush to negative; no membranous |
| 14 | KIF5B-ALK | Archer FusionPlex targeted RNA-seq | No | No | Positive | Granular cytoplasmic with Golgi dot-like accentuation; no membranous |
| 15 | KIF5B-ALK | Archer FusionPlex targeted RNA-seq | Yes | Yes | Positive | Diffuse cytoplasmic with Golgi dot-like accentuation; no membranous |
| 16 | KIF5B-ALK | Archer FusionPlex targeted RNA-seq | No | No | Positive | Granular cytoplasmic with rare Golgi dot-like accentuation; no membranous |
| Group 2: neurological involvement | ||||||
| 17 | KIF5B-ALK | FoundationOne Heme targeted RNA-seq | No | No | Positive | Granular cytoplasmic with Golgi dot-like accentuation in some cells; no membranous |
| 18 | ALK-FISH+ | FISH | No | No | Positive | Diffuse cytoplasmic with Golgi dot-like accentuation; membranous |
| 19 | KIF5B-ALK | Archer FusionPlex targeted RNA-seq | No | No | Positive | Diffuse cytoplasmic; no membranous |
| 20 | KIF5B-ALK | FoundationOne Heme targeted RNA-seq | No | No | Focal, weak | Focal, weak diffuse cytoplasmic with Golgi dot-like accentuation; no membranous |
| 21 | KIF5B-ALK | Archer FusionPlex targeted RNA-seq | Yes | Yes | Positive | Diffuse cytoplasmic with Golgi dot-like accentuation; no membranous |
| 22 | KIF5B-ALK | Whole transcriptome RNA-seq | No | No | Positive | Diffuse granular cytoplasmic; no membranous |
| 23 | KIF5B-ALK | Whole transcriptome RNA-seq | No | No | Positive | Diffuse granular cytoplasmic; no membranous |
| 24 | ALK-FISH+ | FISH | Yes* | No | Positive | Diffuse granular cytoplasmic; no membranous |
| 25 | KIF5B-ALK | Archer FusionPlex targeted RNA-seq | Yes | Yes abundant | Positive | Diffuse cytoplasmic with Golgi dot-like accentuation; membranous |
| 26 | KIF5B-ALK | Ion Ampliseq RNA Fusion Lung Cancer targeted RNA-seq; whole transcriptome RNA-seq | No | Yes abundant | Positive | Diffuse cytoplasmic with Golgi dot-like accentuation; no membranous |
| 27 | KIF5B-ALK | Archer FusionPlex targeted RNA-seq | Yes | No | Positive | Diffuse granular cytoplasmic; no membranous |
| 28 | KIF5B-ALK | FoundationOne Heme targeted RNA-seq | No | No | Focal, weak | Variable focal, weak cytoplasmic to negative; no membranous |
| Group 2: nonneurological involvement | ||||||
| 29 | KIF5B-ALK | Archer FusionPlex targeted RNA-seq | No | No | Positive | Diffuse cytoplasmic with Golgi dot-like accentuation; no membranous |
| 30 | KIF5B-ALK | Oncomine Comprehensive Assay Plus targeted RNA-seq | No | No | Positive | Diffuse granular cytoplasmic with Golgi dot-like accentuation; no membranous |
| 31 | KIF5B-ALK | Archer FusionPlex targeted RNA-seq | No | No | Positive | Diffuse granular cytoplasmic; no membranous |
| 32 | KIF5B-ALK | Whole transcriptome RNA-seq | No | No | Positive | Diffuse dark cytoplasmic; subset possibly nuclear; no membranous (clone 5A4, lung staining protocol). Focal/Golgi dot-like cytoplasmic; no nuclear or membranous (clone 5A4, lymphoma staining protocol) |
| 33 | KIF5B-ALK | Illumina Trusight targeted RNA-seq | No | Yes | Positive | Diffuse light granular cytoplasmic with dark Golgi-dot like accentuation; no membranous |
| 34 | KIF5B-ALK | Illumina Trusight targeted RNA-seq | No | No | Exclusive dot-like | Golgi dot-like cytoplasmic only (focal weak); no membranous |
| 35 | KIF5B-ALK | Archer FusionPlex targeted RNA-seq | No | No | Positive | Diffuse granular cytoplasmic with Golgi dot-like accentuation; no membranous |
| 36 | KIF5B-ALK | FoundationOne Heme targeted RNA-seq | No | No | Exclusive dot-like | Golgi dot-like cytoplasmic only (focal weak); no membranous |
| 37 | KIF5B-ALK | Archer FusionPlex targeted RNA-seq | Yes | Yes | Positive | Diffuse cytoplasmic with Golgi dot-like accentuation; membranous |
| 38 | EML4-ALK | Archer FusionPlex targeted RNA-seq | Yes | No | Positive | Diffuse granular cytoplasmic; membranous |
| 39 | DCTN1-ALK | Archer FusionPlex targeted RNA-seq | Yes | Yes abundant | Positive | Diffuse cytoplasmic with Golgi dot-like accentuation; membranous |
| No. | ALK rearrangement | Analysis method(s) | Classic XG-like | Emperipolesis | ALK IHC | |
|---|---|---|---|---|---|---|
| Staining | Staining pattern | |||||
| Group 1A | ||||||
| 1 | Not confirmed | ALK-FISH negative; KIF5B-ALK RT-PCR negative; no RNA-seq (insufficient material) | No | Yes | Positive | Light cytoplasmic; membranous |
| 2 | ALK-FISH+ | FISH | No | Yes abundant | Positive | Diffuse cytoplasmic; membranous |
| 3 | KIF5B-ALK | Archer FusionPlex targeted RNA-seq | No | No | Positive | Granular cytoplasmic with focal Golgi dot-like accentuation; no membranous |
| 4 | Not confirmed | ALK-FISH negative; no RNA-seq (insufficient material) | Yes | Yes abundant | Positive | Scant cytoplasmic; strong membranous |
| 5 | ALK-FISH+ | FISH | Yes | Yes | Positive | Weak cytoplasmic; strong membranous |
| 6 | CLTC-ALK | Archer FusionPlex targeted RNA-seq | No | Yes | Positive | Diffuse dark cytoplasmic (clone 5A4) to more light granular cytoplasmic (clone ALK1); membranous (both) |
| Group 1B | ||||||
| 7 | TPM3-ALK | Archer FusionPlex targeted RNA-seq | Yes | Yes | Positive | Diffuse cytoplasmic with Golgi dot-like accentuation; membranous |
| 8 | ALK-FISH+ | FISH | No | No | Positive | Diffuse granular cytoplasmic with rare Golgi dot-like accentuation; no membranous |
| 9 | KIF5B-ALK | Illumina Childhood Cancer targeted RNA-seq | No | No | Weak | Very weak diffuse cytoplasmic; no membranous |
| 10 | KIF5B-ALK | Illumina Trusight targeted RNA-seq | No | No | Positive | Diffuse dark cytoplasmic (clone D5F3) to more light, granular cytoplasmic with Golgi dot-like accentuation (clone ALK1); no membranous |
| 11 | KIF5B-ALK | Archer FusionPlex targeted RNA-seq | No | No | Positive | Diffuse cytoplasmic; no membranous |
| 12 | KIF5B-ALK | KIF5B-ALK RT-PCT positive | No | No | Exclusive dot-like | Golgi dot-like cytoplasmic only; no membranous |
| 13 | TFG-ALK | Archer FusionPlex targeted RNA-seq | Yes | No | Focal, weak | Very weak cytoplasmic blush to negative; no membranous |
| 14 | KIF5B-ALK | Archer FusionPlex targeted RNA-seq | No | No | Positive | Granular cytoplasmic with Golgi dot-like accentuation; no membranous |
| 15 | KIF5B-ALK | Archer FusionPlex targeted RNA-seq | Yes | Yes | Positive | Diffuse cytoplasmic with Golgi dot-like accentuation; no membranous |
| 16 | KIF5B-ALK | Archer FusionPlex targeted RNA-seq | No | No | Positive | Granular cytoplasmic with rare Golgi dot-like accentuation; no membranous |
| Group 2: neurological involvement | ||||||
| 17 | KIF5B-ALK | FoundationOne Heme targeted RNA-seq | No | No | Positive | Granular cytoplasmic with Golgi dot-like accentuation in some cells; no membranous |
| 18 | ALK-FISH+ | FISH | No | No | Positive | Diffuse cytoplasmic with Golgi dot-like accentuation; membranous |
| 19 | KIF5B-ALK | Archer FusionPlex targeted RNA-seq | No | No | Positive | Diffuse cytoplasmic; no membranous |
| 20 | KIF5B-ALK | FoundationOne Heme targeted RNA-seq | No | No | Focal, weak | Focal, weak diffuse cytoplasmic with Golgi dot-like accentuation; no membranous |
| 21 | KIF5B-ALK | Archer FusionPlex targeted RNA-seq | Yes | Yes | Positive | Diffuse cytoplasmic with Golgi dot-like accentuation; no membranous |
| 22 | KIF5B-ALK | Whole transcriptome RNA-seq | No | No | Positive | Diffuse granular cytoplasmic; no membranous |
| 23 | KIF5B-ALK | Whole transcriptome RNA-seq | No | No | Positive | Diffuse granular cytoplasmic; no membranous |
| 24 | ALK-FISH+ | FISH | Yes* | No | Positive | Diffuse granular cytoplasmic; no membranous |
| 25 | KIF5B-ALK | Archer FusionPlex targeted RNA-seq | Yes | Yes abundant | Positive | Diffuse cytoplasmic with Golgi dot-like accentuation; membranous |
| 26 | KIF5B-ALK | Ion Ampliseq RNA Fusion Lung Cancer targeted RNA-seq; whole transcriptome RNA-seq | No | Yes abundant | Positive | Diffuse cytoplasmic with Golgi dot-like accentuation; no membranous |
| 27 | KIF5B-ALK | Archer FusionPlex targeted RNA-seq | Yes | No | Positive | Diffuse granular cytoplasmic; no membranous |
| 28 | KIF5B-ALK | FoundationOne Heme targeted RNA-seq | No | No | Focal, weak | Variable focal, weak cytoplasmic to negative; no membranous |
| Group 2: nonneurological involvement | ||||||
| 29 | KIF5B-ALK | Archer FusionPlex targeted RNA-seq | No | No | Positive | Diffuse cytoplasmic with Golgi dot-like accentuation; no membranous |
| 30 | KIF5B-ALK | Oncomine Comprehensive Assay Plus targeted RNA-seq | No | No | Positive | Diffuse granular cytoplasmic with Golgi dot-like accentuation; no membranous |
| 31 | KIF5B-ALK | Archer FusionPlex targeted RNA-seq | No | No | Positive | Diffuse granular cytoplasmic; no membranous |
| 32 | KIF5B-ALK | Whole transcriptome RNA-seq | No | No | Positive | Diffuse dark cytoplasmic; subset possibly nuclear; no membranous (clone 5A4, lung staining protocol). Focal/Golgi dot-like cytoplasmic; no nuclear or membranous (clone 5A4, lymphoma staining protocol) |
| 33 | KIF5B-ALK | Illumina Trusight targeted RNA-seq | No | Yes | Positive | Diffuse light granular cytoplasmic with dark Golgi-dot like accentuation; no membranous |
| 34 | KIF5B-ALK | Illumina Trusight targeted RNA-seq | No | No | Exclusive dot-like | Golgi dot-like cytoplasmic only (focal weak); no membranous |
| 35 | KIF5B-ALK | Archer FusionPlex targeted RNA-seq | No | No | Positive | Diffuse granular cytoplasmic with Golgi dot-like accentuation; no membranous |
| 36 | KIF5B-ALK | FoundationOne Heme targeted RNA-seq | No | No | Exclusive dot-like | Golgi dot-like cytoplasmic only (focal weak); no membranous |
| 37 | KIF5B-ALK | Archer FusionPlex targeted RNA-seq | Yes | Yes | Positive | Diffuse cytoplasmic with Golgi dot-like accentuation; membranous |
| 38 | EML4-ALK | Archer FusionPlex targeted RNA-seq | Yes | No | Positive | Diffuse granular cytoplasmic; membranous |
| 39 | DCTN1-ALK | Archer FusionPlex targeted RNA-seq | Yes | Yes abundant | Positive | Diffuse cytoplasmic with Golgi dot-like accentuation; membranous |
RT-PCR, reverse transcription polymerase chain reaction; XG, xanthogranuloma.
Multiple areas with xanthomatous histiocytes and Touton giant cells were observed; however, a significant part consisted of an infiltration of spindly cells.
Molecular pathologic features
ALK rearrangements were confirmed in 37/39 patients (Table 1). In 2 infants from Group 1A with classic systemic disease, similar to the cases described by Chan et al,29 no ALK rearrangements were detected by FISH; however, the material was insufficient for comprehensive analysis by RNA-seq. KIF5B was the most common ALK fusion partner and identified in 27 cases, with exon 24 of KIF5B fused to exon 20 of ALK in 25/27 cases (supplemental Table 3). In addition, CLTC-ALK, TPM3-ALK, TFG-ALK, EML4-ALK, and DCTN1-ALK fusions were detected in single cases (supplemental Figure 1).
Atypical ALK-rearranged histiocyte-rich tumors: a potential diagnostic pitfall
Three cases were identified without macrophage/histiocyte marker expression by the ALK+ tumor cells but with prominent intermixed CD163-expressing (reactive) macrophages (supplemental Figure 2), which can be mistaken for ALK-positive histiocytosis. Moreover, by hematoxylin and eosin stain, the tumor cells often had similar morphology as those seen in ALK-positive histiocytosis, consisting of plump mononucleated cells with eosinophilic cytoplasm. In addition, the lesional cells demonstrated p-ERK and/or Cyclin D1 expression (supplemental Figure 2). Despite extensive immunohistochemical and molecular pathologic analysis, no definitive diagnosis could be made for the 3 cases. The ALK+ cells lacked immunoreactivity for many lineage markers, including CD30 and smooth muscle actin (supplemental Table 1). Moreover, clonality assessment of T-cell receptor and Ig gene rearrangements did not reveal high monoclonal peaks (supplemental Methods), arguing against a type of lymphoma. CD34 immunostains did reveal a remarkable nested, endocrinoid to hemangiopericytoma-like pattern with abundant blood capillaries in all 3 cases (supplemental Figure 2). None had the KIF5B-ALK fusion but rather harbored ALK fusions involving EML4 or SQSTM1.
Clinically, 1 of the 3 patients was an adult with Group 1B–like multisystemic disease involving the CNS, left lung, and bone (Case A1; supplemental Figure 3A-D). This patient initially had an objective response to ALK inhibition but then demonstrated disease progression and switched to chemotherapy (Figure 7; Table 3). The remaining 2 cases had Group 2–like single-system disease with localized soft tissue tumors. Both were treated with surgery; however, 1 relapsed with subcutaneous and liver metastases (Case A2; supplemental Figure 3E-F) and received systemic treatment with ALK inhibition with stable disease after 3 months of crizotinib.
ALK-immunoreactive histiocytoses without ALK rearrangements
Ten patients were identified with histiocytoses demonstrating variable ALK immunoreactivity (supplemental Figure 4) but no ALK rearrangement by RNA-seq. Both clinically and histologically, these cases represented the full spectrum of histiocytic neoplasms, from L-, C-, and R- to M-group (supplemental Table 4).1 When compared with ALK-positive histiocytoses, they more often showed nuclear ALK staining. Other MAPK activating somatic mutations were identified in 5/10 patients.
Discussion
We present here a collaborative international study of ALK-positive histiocytosis with detailed clinicopathologic data of 39 cases, including 37 cases with confirmed ALK rearrangements. This study defines a new clinicopathologic spectrum and highlights frequent neurologic involvement. Histology included classic xanthogranuloma features in almost one-third of patients, whereas the majority displayed a more densely cellular, monomorphic appearance without lipidized histiocytes. We affirm the frequent occurrence of KIF5B-ALK fusions43 and expand the molecular spectrum by describing single cases with CLTC-ALK, TPM3-ALK, TFG-ALK, EML4-ALK, or DCTN1-ALK fusions. Finally, we show dramatic and durable responses in 11/11 patients treated with ALK inhibition (Figure 7), 10 with neurologic involvement.
We noted an apparent predilection for females in our cohort of ALK-positive histiocytosis cases, consistent with our overview of the existing literature (supplemental Table 5) and the female predilection observed in ALK-rearranged tumors other than non–small cell lung cancer,50 such as ALK-rearranged inflammatory myofibroblastic tumors.59 This female predilection is in clear contrast to the male predominance in LCH,60,61 ECD,62,63 and JXG,64,65 particularly in BRAF p.V600E–associated CNS lesions with JXG histology.28
Our Group 1A patients were similar to the cases originally described by Chan et al.29 In addition to liver, spleen, and hematopoietic involvement, our patients had lesions in the lungs, kidneys, and/or skin. Unlike in LCH,66 in which liver, spleen, and/or hematopoietic involvement constitutes “risk organ” involvement associated with increased mortality, this clinical phenotype in ALK-positive histiocytosis does not necessarily comprise high-risk disease as 2 of 6 patients from Group 1A had spontaneous regression of disease with only supportive care. However, of the 3 other patients with clinical follow-up, 3/3 required second-line systemic therapy and 1 died, underscoring the life-threatening condition that this disease can still represent.29
Other patients with multisystemic ALK-positive histiocytosis (Group 1B) have been variably reported previously (supplemental Table 5), sometimes as ECD or systemic JXG. The nervous system was the most frequently involved organ in our patients from this group, followed by the lungs, bone, liver, skin, and lymph nodes. Although there are similarities between patients from this group and patients with ECD, particularly bilateral bone lesions in the legs, many other stigmata of ECD were not observed. For example, perinephric soft tissue thickening (“hairy kidneys”), periaortic encasement, diabetes insipidus, and right atrial infiltration are frequent in ECD62,67 but were absent in our cohort. Moreover, ECD patients are generally older, with a median age of 55 years,2,63 as opposed to a median age of 14.5 years of our Group 1B patients. Apart from the specific phenotypic entity that is ECD, there exists the constellation of “extracutaneous/systemic JXG,”68,69 referring to the rare disease in childhood with JXG histology involving extracutaneous tissues. In a recent series of “systemic JXG,” associations were observed between specific molecular alterations and clinical phenotypes, with ALK rearrangements overrepresented in female cases and cases with lung involvement.68 These findings are compatible with ours, raising the question whether ALK-positive histiocytosis is distinct from “systemic JXG” without ALK rearrangements. Altogether, although there is overlap in histomorphology and immunophenotype, we believe that sufficient evidence is available for the designation of ALK-positive histiocytosis as a separate histiocytic entity from both ECD and “extracutaneous/systemic JXG” as previously proposed by others.45
The findings in our patients with single-system disease (Group 2) were similar to that which has been observed previously (supplemental Table 5), with the nervous system and skin/soft tissue as common sites of disease and surgical resection often being definitive. We expand the clinical spectrum by describing patients with localized bone involvement. When compared with previously reported cases with single-system disease, our cohort includes more cases with isolated neurologic involvement (52% vs 19%) and more children (87% vs 38%). Given the retrospective nature of our study, we cannot rule out some selection bias influencing the clinical spectrum of the disease in our cohort. For example, our cohort did not include adult female patients with isolated breast masses as recently described by Kashima et al34 and Osako et al.41 Yet, our study did include 2 patients with multisystemic disease including breast masses (Table 3; Figure 3R), supporting the recurrent involvement of this anatomic site.
Eleven patients in our cohort were treated with diverse ALK inhibitors, either as first- or second- or further-line therapy, and sustained objective responses were observed in 11/11 patients (Figure 7). These data corroborate the favorable outcomes of ALK inhibition observed in 7 ALK-positive histiocytosis patients outside of our cohort.33,34,40,43,50,51 Our patients were treated with various ALK inhibitors at variable doses; however, the impression of their efficacy is unequivocal. The responses to ALK inhibition stand in contrast to outcomes with conventional systemic therapy, which conferred an objective response in only 7/13 (54%) patients as first-line therapy and 2/5 (40%) patients as second-line therapy (Table 2). However, given that this is a retrospective study, it remains undefined whether ALK inhibition should be implemented as first-line treatment or when disease is refractory to conventional therapies. Also, our study does not address the optimal treatment regimen or duration for ALK inhibition, which is an enduring question for targeted therapy of histiocytosis more broadly, as cessation of BRAF inhibition in histiocytosis patients has been shown to lead to early relapses in most cases.22,26,70
As illustrated by the 3 “atypical ALK-rearranged histiocyte-rich tumors,” stringent histopathologic assessment is essential before making a diagnosis of ALK-positive histiocytosis. Diagnosis requires diffuse expression of macrophage/histiocyte markers, preferably CD163, by the lesional ALK+ tumor cells. ALK-rearranged tumors with prominent intermixed histiocytic infiltrates disparate from ALK+ cells without immunoreactivity for macrophage/histiocyte markers are a potential diagnostic pitfall and should be assessed for the possibility of representing another ALK-rearranged entity, such as inflammatory myofibroblastic tumors, which typically do not harbor KIF5B-ALK fusions.71,72 Future studies may elucidate whether “atypical ALK-rearranged histiocyte-rich tumors” without characteristics of established (ALK-rearranged) entities comprise a new entity. In all 3 of our cases, the CD34 immunostain revealed a remarkable nested pattern with abundant blood capillaries (supplemental Figure 2), which was also observed in a case previously reported as ALK+histiocytosis with unusual morphology and a TRIM33-ALK fusion.32 Clinically, this patient had a large mesenteric tumor, highly similar to Case A2 from our study (supplemental Figure 3E-F). Finally, the monomorphic dense infiltration of histiocytes seen in the majority of our ALK-positive histiocytosis cases should be distinguished from epithelioid fibrous histiocytoma.73
Supported by our 10 excluded histiocytosis cases demonstrating variable ALK immunoreactivity but no ALK rearrangement by RNA-seq, molecular confirmation of ALK rearrangement should be performed whenever possible to confirm the diagnosis of ALK-positive histiocytosis. This is important because cases with ALK staining but without ALK rearrangements may not benefit from ALK inhibition.74,75 ALK IHC should particularly be applied in the diagnostic workup of infants with classic multisystemic disease and patients with tumorous lesions in the nervous system, lungs, bone, and/or soft tissue with non-LCH histology. Yet, pathologists should be aware of the variable staining intensity and pattern, with variations further compounded by the ALK clone and protocol used. Comprehensive molecular analysis (ie, RNA-seq) should be performed in ALK IHC–negative cases when the detection of an ALK rearrangement could have treatment consequences as we noted in 7 confirmed ALK-rearranged cases weak or exclusive Golgi dot-like cytoplasmic ALK staining that could be misinterpreted as negative.
Overall, this study defines a new clinicopathologic spectrum of ALK-positive histiocytosis with frequent neurologic involvement and durable responses to ALK inhibition. The findings support the recognition of ALK-positive histiocytosis as a distinct entity from both ECD and (extracutaneous/systemic) JXG. Comprehensive genomic analysis of patients with histiocytic neoplasms is now a necessary adjunct to detailed morphologic and immunophenotypic assessment as this may directly affect disease classification and therapeutic decision making.
Acknowledgments
The authors acknowledge the International Rare Histiocytic Disorders Registry (IRHDR, ClinicalTrial.gov number NCT02285582), which included some patients described in this study. The authors thank Chris Woods (Lead Analyst, Imaging Informatics) and the Research Pathology Laboratory at Cincinnati Children’s Hospital Medical Center, John DeCoteau (molecular pathologist) at University of Saskatchewan, Sven van Kempen (senior laboratory technician) and Lennart Kester (clinical molecular biologist in pathology in training) at Princess Máxima Center for Pediatric Oncology, and Demi van Egmond (laboratory technician), Tom van Wezel (clinical molecular biologist in pathology), and Arjen Cleven (pathologist) at Leiden University Medical Center for technical assistance. The authors thank Els Wauters (pulmonologist and respiratory oncologist) at University Hospitals Leuven, Andreas Rosenwald (pathologist) at the Institute of Pathology of the University of Würzburg, and Bipin Mathew (pathologist) at Leeds Teaching Hospitals for clinical care and/or diagnostics of patients described in this study. The authors thank Les Laboratoires Servier for allowing the use of their medical illustrations (Servier Medical Art, https://smart.servier.com) in the visual abstract. Figure 6 was created with BioRender.com.
P.G.K. received a grant from Leiden University Medical Center. A.G.S.v.H. is supported by Stichting 1000 kaarsjes voor Juultje. E.L.D. is supported by the National Institutes of Health/National Cancer Institute Core Grant (P30 CA008748), National Cancer Institute (R37 CA259260-01), the Frame Fund, Applebaum Foundation, and Joy Family West Foundation. J.-F.E. is supported by the Programme de Recherche Translationnelle sur le Cancer from the French National Cancer Institute (PRT-K19-143).
Authorship
Contribution: P.G.K., J.P., E.L.D., and J.-F.E. conceived the study and drafted the manuscript; J.P. and J.-F.E. performed the central pathology review and provided histologic images; P.G.K., J.P., B.H.D., Z.H.-R., L.H.-J., and J.-F.E. performed additional immunohistochemical and/or molecular analyses; P.G.K. made the figures; A.G.S.v.H. participated in data discussions; A.G.S.v.H. and O.A. provided supervision; and all authors contributed cases, and read, revised, and approved the manuscript (meeting the International Committee of Medical Journal Editors criteria for authorship).
Conflict-of-interest disclosure: E.L.D. discloses unpaid editorial support from Pfizer Inc and paid advisory board membership from Day One Biopharmaceuticals outside the submitted work. M.E. discloses honoraries and travel grants by Jazz Pharmaceuticals, Merck Sharp & Dohme, and Amgen. V.P. discloses giving lectures supported by Sanofi, R-farma, and Roche and participation in an expert board supported by Novartis. J.L.H. is a consultant to Aadi Biosciences and TRACON Pharmaceuticals outside the scope of the manuscript. The remaining authors declare no competing financial interests.
Correspondence: Jennifer Picarsic, Division of Pathology, Cincinnati Children’s Hospital Medical Center, 3333 Burnet Ave, ML 1035, Cincinnati, OH 45229; e-mail: jennifer.picarsic@cchmc.org; Eli L. Diamond, Department of Neurology, Memorial Sloan-Kettering Cancer Center, 160 East 53rd St, 2nd Floor Neurology, New York, NY 10022; e-mail: diamone1@mskcc.org; and Jean-François Emile, EA4340 Laboratory and Department of Pathology, Ambroise Paré Hospital, 9 Avenue Charles de Gaulle, 92104 Boulogne, France; e-mail: jean-francois.emile@uvsq.fr.
Original data are available from the corresponding authors upon reasonable request.
There is a Blood Commentary on this article in this issue.
The online version of this article contains a data supplement.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal