

Abstract
Polycythemia vera (PV) is a Philadelphia chromosome-negative myeloproliferative neoplasm driven by the JAK2 V617F (or rarely exon 12) mutation. Its natural history can extend over a few decades, and therefore treatment planning is predicated on continual reassessment of traditional risk features (age, prior thrombosis) to evaluate the need for cytoreduction besides foundational therapy with low-dose aspirin and stringent phlebotomy. Shorter- and longer-term patient goals should be considered in light of several variables such as comorbid conditions (especially cardiovascular risk factors), disease symptoms, and the risk-benefit profile of available drugs. While hydroxyurea has been the pro forma choice of cytoreduction for many practitioners over the last half-century, the more recent regulatory approvals of ruxolitinib and ropeginterferon-alfa-2b, based on phase 3 randomized trials, highlight an expanding portfolio of active drugs. Obtaining high-level evidence for short-term clinical trial endpoints such as hematocrit control, symptom burden/quality of life, splenomegaly, and JAK2 V617F allele burden lies within the timeline of most studies. However, in many cases, it may not be possible to adequately power trials to capture significant differences in the typically low event rates of thrombosis as well as longer-horizon endpoints such as evolution to myelofibrosis and acute myeloid leukemia and survival. This Perspective highlights the challenges of addressing these data gaps and outstanding questions in the emerging treatment landscape of PV.
Introduction
When results from a phase III trial become available in an orphan disease, it provides an opportunity to take stock of the current treatment landscape. The MPD-RC 112 randomized trial of hydroxyurea (HU) vs peginterferon-α-2a (PEG-IFN; PEGASYS) in high-risk polycythemia vera (PV) and essential thrombocythemia (ET) reported by Mascarenhas and colleagues1 in this issue is no exception. While the hypotheses driving such studies attempt to provide high-level evidence for unanswered treatment questions, each trial exists in a historical framework that reflects diverging interpretations of the body of accumulated data, bookended by generally agreed-upon facts and lingering myths.2-4 This is certainly emblematic of PV.
Of the many questions that animate the PV field, a key one is the comparative safety and efficacy of HU vs pegylated forms of IFN-α in high-risk disease, traditionally defined as age ≥60 years and/or a history of thrombosis.5,6 Debates about these drugs generally center on the following points: (1) HU’s effects on blood counts are felt to be more cosmetic, with mixed evidence regarding effects on hematopoietic stem and progenitor cells7-10 and no available data to support an impact on disease modification; and (2) persistent concerns remain about the drug’s leukemogenicity despite not having being borne out by several studies.11-15 This is particularly relevant in a disease whose arc already bends toward myelofibrosis (MF) and acute myeloid leukemia (AML), with respective rates of ∼10% to 20% and ∼5% to 10% over 10 to 20 years.15
IFNs exert selective effects on the clonal outgrowth of mutant myeloproliferative neoplasm (MPN) hematopoietic stem and progenitor cells,16-19 with several studies now consistently demonstrating partial and, less frequently, complete molecular responses of JAK2 V617F.20-25 Where the attractiveness of reducing the JAK2 V617F allele clonal burden and, by extension, the promise of altering PV natural history remain the “rose” of IFN therapy, the prickly “thorn” has been the drug’s reputation of poor tolerability (eg, flu-like side effects; liver function abnormalities, endocrine/thyroid disorders, autoimmune phenomena, mood lability). Phase 1/2 studies of pegylated IFNs have reported discontinuation rates in the range of ∼15% to 30% (not all related to adverse events).20-25 While pegylation has improved the drug’s toxicity profile, lingering concerns about tolerability contribute to IFN hesitancy by physicians and patients.
Phase 2/3 randomized trials of IFNs
With this background on IFNs, the MPD-RC 112 trial randomized 168 patients with treatment-naïve, high-risk PV (n = 87) and ET (n = 81) to HU vs PEG-IFN.1 The median duration of treatment/median weekly dose was 81 weeks/6708 mg in the HU arm and 94.6 weeks/89.4 μg in the PEG-IFN arm. The primary endpoint of complete response (CR), defined by the EuropeanLeukemia Net (ELN) as a composite of normalization of blood counts and resolution of splenomegaly and disease-related symptoms,26 was achieved in 37% and 35% of patients treated with HU and PEG-IFN, respectively (P = .80). In the PV cohort, the 12-month CR rates were similar, 30% (HU) vs 28% (PEG-IFN), as well as the rates of phlebotomy. However, hematocrit control (<45%, without phlebotomy) was achieved in 43% and 65% of patients with PV receiving HU and PEG-IFN, respectively (P = .04), suggesting superiority of the latter agent in controlling blood cell counts. Statistically significant differences in the CR rates between the 2 drugs did not emerge after 24 and 36 months of follow-up.1
The finding that the bone marrow histopathologic responses were higher with HU compared with PEG-IFN came as a surprise to MPN investigators. Marrow responses were more frequent in ET vs PV, in which best histopathologic responses were recorded in only 12% of patients at 12 months. A dose-dependent effect on histopathologic responses was observed with HU but not with PEG-IFN, which may indicate that myelosuppression rather than a disease-modifying effect of HU is contributing to this differential response. Also, these marrow responses stand in contrast to the trajectory of molecular responses, in which the median JAK2 V617F variant allele frequency (VAF) decreased steadily through month 24 on PEG-IFN but increased after 12 months in the HU cohort.1 These observations, in addition to the lack of central pathology adjudication, have invited skepticism about these histopathology results. Overall, grade 3/4 adverse events were more common with PEG-IFN compared with HU (46% vs 28%). Ultimately, the MPD-RC 112 trial was handicapped by sponsor withdrawal of PEG-IFN supply, and therefore, its statistical power, tethered to a planned accrual of 300 patients, was not realized. Despite the obstacles that limited enrollment and duration of follow-up, it can be reasonably concluded that HU and PEG-IFN elicit similar shorter-term control of blood counts.
MPD-RC 112 has drawn inevitable comparisons to the contemporaneous 12-month PROUD PV and 48-month extension CONTINUOUS-PV studies of roPEG-IFN-α-2b (roPEG-IFN; BESREMi) vs HU.27,28 RoPEG-IFN is a monopegylated IFN consisting of a single positional isomer that permits extended dosing every 2 weeks and monthly administration during maintenance.29 Entry criteria for PROUD-PV were not defined by the aforementioned traditional high-risk PV criteria; instead, individuals without prior cytoreduction but in need of cytoreductive therapy were eligible or if they were treated with HU for <3 years and had no prior CR or were intolerant to HU according to ELN criteria.26-28 At the 60-month interim analysis, the complete hematologic response (CHR) rates were 56% and 44% in the roPEG-IFN and HU arms, respectively (P = .0974), in which discontinued patients were counted as nonresponders. When last counts before discontinuation were carried forward, differences in the CHR rates remained statistically significant from month 24 through month 60. In the fifth year of treatment, 81.8% of patients in the roPEG-IFN arm vs 63.2% in the HU arm demonstrated freedom from phlebotomy. The dynamics of molecular response that emerged during the first 24 months of MPD-RC 112 mirror the results of CONTINUATION PV; from years 2 to 5, a further significant reduction in JAK2 V617F allele burden and an increase in percentage of molecular responders was noted in patients treated with roPEG-IFN, whereas the opposite temporal trend occurred with HU. These data provide a useful reminder about IFNs: hematologic and (especially) molecular responses may be slow to emerge, and PV should recall the adage “patience is a virtue.” The drug was generally well tolerated. At a median follow-up of 36 months, the rates of discontinuation due to adverse events were 4% and 8% in the HU and RoPEG-IFN arms, respectively. These data supported the approval of roPEG-IFN by the European Medicines Agency in 2019 for adult patients with PV without splenomegaly; the US Food and Drug Administration granted approval in 2021 for the same patient population but agnostic to the presence of splenomegaly.
RoPEG-IFN was also evaluated in a randomized phase II trial that compared standard therapy (phlebotomy plus low-dose aspirin) alone vs standard therapy plus roPEG-IFN in low-risk patients with PV.30 The study authors provided several rationales for use of IFN in low-risk disease that help frame potential clinical issues in this patient population: (1) the CYTO-PV trial31 showed that maintenance of a hematocrit <45% is associated with a 4-fold decrease in major thrombosis, but evidence is lacking for phlebotomy alone being able to sufficiently maintain the hematocrit at this target level; (2) low-risk patients with PV exhibit a rate of major arterial and venous events (2% patient-years) that is 2- to 3-fold higher than the general population with or without multiple risk factors32-35; (3) phlebotomy alone does not control leukocytosis, which has been identified as an associated risk factor for thrombosis in some (but not all) studies36-38; and (4) the impact of repeated phlebotomy can impact quality of life (QOL)39 and produce symptoms of iron deficiency (eg, fatigue, headaches, dizziness, restless legs).
The study randomized 127 patients to standard therapy (n = 63) and the experimental arm of roPEG-IFN plus standard therapy (n = 64).30 At 12 months, the experimental arm exhibited superior rates of target hematocrit control, reduced frequency of phlebotomy, increased ferritin blood concentrations, and normalization of the white blood cell (WBC) and platelet counts. Only 1 thrombotic event occurred in the standard therapy arm. Although the total number of adverse events was significantly higher with experimental therapy, a low rate of grade 3 or higher adverse events was recorded and was similar between the 2 arms. With the exception of higher symptom scores for fatigue and fever in the experimental group, none of the other symptoms were improved by the phlebotomy-only arm but were all reduced by the addition of roPEG-IFN. This positive trial has implications on how to approach treatment in low-risk patients with PV in whom cytoreduction (especially HU) is generally not recommended except in select circumstances.5,6
Ruxolitinib
The experience with ruxolitinib (RUX) in MF40,41 showed that higher grade anemia and thrombocytopenia are fairly common and relate to on-target effects of JAK-STAT pathway inhibition. These drug-related hematologic toxicities are unwelcome in MF but in PV can be tolerated, and better yet, exploited, when JAK2 V617F-driven increases in red blood cell mass, and often other blood lineages, define the blood picture. In addition, the well-established improvements in splenomegaly and symptoms/QOL with RUX in MF40-42 are also relevant clinical goals in PV. However, symptomatic splenomegaly in PV does not exhibit the clinical burden that it does in advanced MF.
The RESPONSE trial43 evaluated RUX in patients with PV who were resistant and/or intolerant to HU as defined by ELN criteria.44 Resistance to HU has been associated with a 5.6-fold increase in risk of death and 6.8-fold increase in risk of transformation.45 RESPONSE randomized 222 phlebotomy-dependent patients with PV with splenomegaly in a 1:1 ratio to RUX (n = 110) and to standard therapy (n = 112), which comprised HU (59%), IFN (12%), anagrelide (7%), immunomodulatory therapies (5%), and no treatment (15%). RUX was initially dosed at 10 mg twice daily and titrated to achieve the primary endpoint while avoiding ≥grade 2 cytopenias. The composite primary endpoint of hematocrit control and ≥35% reduction in spleen volume by week 32 was achieved in 21% of patients treated with RUX vs 1% on standard therapy. Hematocrit control/CHR were achieved in 60%/24% vs 20%/9% of patients receiving RUX vs the standard arm. In addition, ≥35% spleen volume reduction was achieved in 38% vs 1% of these groups, respectively. A >50% reduction in total symptom score occurred in 49% of patients treated with RUX vs only 5% of patients treated with standard therapy. Six cases of thromboembolism occurred in the standard therapy arm vs 1 in the RUX cohort. Herpes zoster infection was recorded in 6% of patients treated with RUX but was not observed in the standard treatment group.
At 5-year follow-up of RESPONSE,46 the probabilities of maintaining the primary composite endpoint and complete clinicohematologic remission were 74% and 55%, respectively. Rates of thromboembolism were 1.2 per 100 patient-years in the RUX group, 2.7 per 100 patient-years in crossover patients, and 8.2 per 100 patient-years in the standard therapy arm, but the trial was not powered for these endpoints. Survival, based on an intention-to-treat analysis, was similar between the 2 arms. The rate of RUX discontinuation was low at 15%, but rates of herpes zoster infection and nonmelanoma skin cancer (including without prior HU exposure) were more common in the patients treated with RUX. These data supported the regulatory approval of RUX as a second-line option for PV in adults who have had an inadequate response to, or intolerance of, HU. The randomized, open-label, phase 3b, RESPONSE-2 trial was conducted in a similar population of patients with PV but without splenomegaly.47 The study corroborated its sister trial’s result by demonstrating hematocrit control in 62% of patients treated with RUX and 19% in the standard therapy arm. Finally, the randomized, double-blind, phase 3b RELIEF trial evaluated PV-related symptoms in patients who were well controlled with a stable dose of HU but had persistent symptoms.48 A nonsignificant trend in improved PV-related symptoms was observed with RUX compared with HU. The authors surmised that these results may be explained by a larger than expected number of patients maintained on HU who exhibited symptom improvement.
A summary of the outcomes of the randomized phase 2 and 3 trials of pegylated IFNs and RUX is detailed in Table 1.
The thrombosis endpoint
Thrombosis is an ever-present concern in PV, and reduction of vascular events is a primary treatment goal to reduce morbidity and mortality5,6; in 1 large series, thrombosis accounted for 20% of known causes of death.15 In a meta-analysis that pooled results across 13 436 patients with MPN, prevalence of thrombosis was particularly elevated at initial diagnosis, (28.6%, 20.7%, and 9.5% of newly diagnosed patients with PV, ET, and MF, respectively), likely reflecting uncontrolled blood counts.49,50 Another meta-analysis of >9000 patients with MPN that compared thrombosis rates to age- and sex-matched controls found a hazard ratio of 4.8 three months after diagnosis, decreasing to 1.8 five years after diagnosis.49,51 Gender differences in patterns of thrombosis were observed in patients enrolled in the ECLAP study. For example, men exhibited higher rates of myocardial infarction and peripheral arterial disease, whereas women experienced higher rates of venous thrombosis, particularly splanchnic vein thrombosis, which was a common presenting manifestation of PV.15,52
The conventional prognostic system for PV is anchored to age and history of thrombosis. Another prognostic risk model that stratifies patients into 3 risk groups with median overall survivals of 10.9, 18.9, and 27.8 years assigns points for 2 age ranges (≥67 years vs 57-66 years), leukocyte count (≥15 × 109/L), and history of venous thrombosis.15 In keeping with prior data,53 a recent multivariable analysis confirmed a JAK2 V617F VAF >50% as an additional independent risk factor for future venous (but not arterial) thrombosis.54 However, diabetes, hyperlipidemia, and previous arterial thrombosis were identified as independent predictors of future arterial thrombosis.54 In an analysis of low-risk patients with PV, thrombosis-free survival was 34% vs 66% in individuals with and without hypertension.55 Cardiovascular risk factors are not currently included in PV risk classification (in contrast to the International Prognostic Score for Thrombosis in ET),56 but guidelines recommend their optimization regardless of PV risk group.5,6
In MPNs, no correlation has been found between thrombocytosis and risk of thrombosis; instead, risk of bleeding increases at the extremes of platelet count.3,57 Several studies have observed a relationship between leukocytosis and thrombosis in MPNs, including PV.36,57 However, other studies have not confirmed this finding, which may relate to the modeling methods used to analyze the longitudinal trajectories of WBC counts and hazard of thrombosis.38 In the ECLAP study, which demonstrated a 60% risk reduction in vascular events with low-dose aspirin, baseline leukocytosis was identified as an independent risk factor for thrombosis.32,58 In the CYTO-PV trial,31 patients were randomized to different intensities of therapy to maintain the hematocrit <45% vs 45% to 50%. A 4-fold increase in the rate of major thrombosis and cardiovascular death was observed in patients randomized to the higher hematocrit target range. Although platelet counts were similar between the 2 arms, a post-hoc analysis revealed that a higher WBC count persisted in the higher hematocrit arm.37 A multivariable analysis revealed that the risk of thrombosis was increased with a WBC count >7 × 109/L, an association that became statistically significant at a WBC count threshold >11 × 109/L.37 No prospective data exist to answer the question of whether normalizing the WBC count is an effective strategy for reducing the incidence of thrombosis.
The ECLAP32 and CYTO-PV31 studies yielded high-level data for the use of low-dose aspirin and stringent hematocrit control to reduce vascular complications. In addition to these cornerstones of therapy, consensus guidelines recommend cytoreduction in high-risk patients with PV for prevention of thrombosis.5,6 However, apart from data extrapolated from ET,59-62 there are no randomized trial data in PV demonstrating that HU (or other cytoreductives) compared with phlebotomy alone reduce vascular risk. Lower-level evidence from the Polycythemia Vera Study Group protocol 08 showed that HU decreased the incidence of thrombosis compared with a cohort of historical controls treated with phlebotomy alone.63 Barbui and colleagues conducted a propensity-matched analysis of 1042 patients from the ECLAP study treated only with phlebotomy or HU to maintain the hematocrit <45%.64 HU reduced the incidence of fatal and nonfatal cardiovascular events (5.8 vs 3.0 per 100 person-years; P = .002).64 Notably, the excess of mortality and cardiovascular events in the phlebotomy arm was restricted to the high-risk patients with PV and was most striking in individuals failing to achieve the hematocrit target.
The phase 3 randomized trials of PEG-IFN,1 roPEG-IFN,27,28,30 and RUX43,46-48 were not designed for or statistically powered to assess the endpoint of thrombosis rate. Event rates are low, and even with extended follow-up, the absolute number of thrombosis events in both the experimental and controls arms of these trials have typically ranged in the single digits. Shorter-term trial endpoints, including CHR, hematocrit control, freedom from phlebotomy, and changes in JAK2 V617F allele burden, are not considered sufficient surrogates for inferring vascular event rates. In addition, recent data indicate that achieving a response per ELN criteria is not associated with a reduced hazard of thrombosis and therefore, may not be informative in gauging the impact of therapies on this endpoint.65
The assessment gap
The difficulty in generating high-level evidence for the impact of specific cytoreductive agents on thrombosis also highlights the challenge of evaluating longer-term PV endpoints such as transformation to myelofibrosis or AML and overall survival (Figure 1). Conducting randomized trials with 10- to 15-year horizons to capture these endpoints is neither practical nor feasible for all stakeholders (patients, physicians, and biopharma sponsors). Short of randomized trials, lower-level evidence derived from sources such as retrospective analyses,66 real-world observational studies (eg, REVEAL),67,68 and national registries is therefore needed to bridge this “assessment gap” by building a body of evidence that points to a particular therapy’s impact on the natural history of a chronic disease. This is akin to investigative journalism, in which short of a “smoking gun,” multiple corroborating sources are required to build a narrative that, in the words of famed Watergate journalist Carl Bernstein, is the “best obtainable version of the truth.”69

The assessment gap in PV. Clinical trial endpoints such as hematocrit (Hct) control, splenomegaly, symptoms/quality of life (QOL), and JAK2 V617F allele burden are shorter-term clinical trial endpoints for which higher-level evidence has been obtained with randomized trials evaluating aspirin/phlebotomy, hydroxyurea (HU), ruxolitinib (RUX), and pegylated interferons (IFNs). However, endpoints with lower-event rates (eg, thrombosis) or longer-horizon outcomes such as evolution to myelofibrosis (MF) and acute myeloid leukemia (AML), and survival cannot be readily assessed within the timeframe of such trials. Therefore, bridging this data divide in the setting of a chronic myeloid neoplasm such as PV requires reliance on aggregate data from surrogate studies (eg, large retrospective and real-world observational studies and registry analyses). Professional illustration by Ian Baker; figure conceived by J.G.
The assessment gap in PV. Clinical trial endpoints such as hematocrit (Hct) control, splenomegaly, symptoms/quality of life (QOL), and JAK2 V617F allele burden are shorter-term clinical trial endpoints for which higher-level evidence has been obtained with randomized trials evaluating aspirin/phlebotomy, hydroxyurea (HU), ruxolitinib (RUX), and pegylated interferons (IFNs). However, endpoints with lower-event rates (eg, thrombosis) or longer-horizon outcomes such as evolution to myelofibrosis (MF) and acute myeloid leukemia (AML), and survival cannot be readily assessed within the timeframe of such trials. Therefore, bridging this data divide in the setting of a chronic myeloid neoplasm such as PV requires reliance on aggregate data from surrogate studies (eg, large retrospective and real-world observational studies and registry analyses). Professional illustration by Ian Baker; figure conceived by J.G.
Surrogate approaches have been used to study the impact of IFNs, touted as disease-modifying agents, on the longer-term disease course of PV. A single institutional experience of 470 patients compared the endpoints of evolution to myelofibrosis and overall survival between recombinant IFN-α, HU, and phlebotomy alone.70 The median overall survival for IFN-α, HU, and phlebotomy groups was 27.7, 25.9, and 21.3 years, respectively (P < .01). In a multivariable analysis including age, sex, cardiovascular risk factors, and thrombosis history, patients treated with IFN-α had a significantly lower risk of transformation to MF compared with HU and phlebotomy. The IFN-α group also exhibited a lower risk of death compared with the HU (HR = 0.33; P = .01) and phlebotomy-only (HR 0.3; P = .001) cohorts, independent of age.70 HU did not decrease the risk of transformation to MF or mortality. Rates of discontinuation were similar between the 2 drugs.
The same institution used a matched-propensity analysis to compare survival of their PV cohort to patients with PV abstracted from The National Cancer Institute's Surveillance, Epidemiology and End Results (SEER) Program, from 2001 to 2017.71 Their study found that overall survival was almost 11 years longer among their patients with PV compared with the SEER PV cohort, representing a 65% reduction in mortality risk. In comparison with the actuarial overall survival of the US population, survival of the SEER patients with PV was significantly worse (mortality HR 2.47), whereas survival of the institutional cohort was similar (mortality HR 1.15).71 The basis for these differences is not easily known but may reflect the expertise of specialized MPN referral centers and their adherence to treatment guidelines compared with other practice settings.
Symptoms: the patient voice
Development and validation of the myeloproliferative neoplasm symptom assessment form (MPN-SAF)72 and abridged 10-item MPN SAF total symptom score (MPN-SAF TSS; MPN-10)73 by Mesa and colleagues have provided much needed harmonization and quantitative assessment of patient symptom burden and QOL. These measures of patient-reported outcomes have been accepted by the regulatory authorities in the drug review process of registrational trials of MPN drugs (eg, RUX for MF and PV) and are now widely used in clinical practice. TSS-10 has shown significant differences in the symptom burden between MPNs (MF>PV>ET) but also heterogeneity within MPNs.73 For example, in PV, TSS score severity increased across 5 clusters but did not vary by PV risk group or age.74 TSS has been used to try to deconstruct the individual contributions of PV-related variables such as phlebotomy requirements, splenomegaly, and HU use to the symptom burden.75 An additive relationship exists between the number of these features present and the degree of symptomology reported on the TSS. The MPN-SAF has also provided insight into the burden of symptoms and their relationship to depression,76 sexual dysfunction,77 and impact on daily activities and work productivity.78
The MPN Landmark Survey revealed important insights about the misalignment between physician and patient perceptions of symptom burden and treatment goals.79 While both patients and physicians identified fatigue as the single symptom most needing attention, physicians underestimated the proportion of patients with symptomatic PV compared with patients. In addition, among patient respondents, slow/delay progression of the condition was the most important treatment goal whereas physicians indicated prevention of vascular/thrombotic events.79 Better understanding of these gaps between physicians and patients will help foster improved communication about PV-related symptoms and promote more realistic expectations about treatment goals, especially given the lack of available drugs to delay progression or improve survival. This should also encourage more nuanced physician-patient conversations about the efficacy and tolerability of drugs such as IFNs, RUX, and HU based on the individual’s clinical features rather than drawing upon unfiltered information from the internet or other sources.
Novel agents
In recent years, clinical trials have commenced evaluation of novel drugs with different mechanisms of action, including inhibitors of MDM2 (eg, idasanutlin, KRT-232),80-82 LSD1 (bomedemstat; IMG-7289),83 histone deacetylase (givinostat),84 and agents that decrease the availability of iron to the bone marrow erythron. In PV, expanded erythropoiesis, iron deficiency, and increased erythroferrone levels lead to suppression of hepcidin and enhanced availability of iron for erythropoiesis.85 Therefore, efforts have been made to develop therapeutics for patients with PV that promote hepcidin expression or mimic its function. Hepcidin expression is negatively regulated by the transmembrane protease serine 6 (TMPRSS6).86 IONIS-TMPRSS6-LRX is a highly specific and potent antisense oligonucleotide targeting TMPRSS6 mRNA, which results in dose-dependent hepcidin upregulation and is under development for PV.87 In patients with PV who require phlebotomy with or without concurrent cytoreduction, subcutaneous administration of the hepcidin mimetic rusfertide (PTG-300) has demonstrated the ability to eliminate phlebotomy requirements, increase serum ferritin concentrations, and improve symptoms, which may be partly related to iron deficiency.88 A phase 3, randomized, double-blind, placebo- controlled trial of rusfertide vs placebo as add-on to each patient’s ongoing therapy for PV is commencing.89 Agents that modulate iron homeostasis are not expected to exert disease-modifying effects but can potentially address issues that affect patient symptoms and QOL, such as the inconvenience and side effects of phlebotomy and iron deficiency. Over the next decade, efforts will focus on the technical and clinical feasibility of gene editing to correct JAK2 V617F in autologous-transplanted PV HSC.90
Summary and future directions
Treatment of the patient with PV reflects several core concepts: (1) risk stratification, (2) adherence to well-evidenced foundational therapy (low-dose aspirin and stringent hematocrit control) to reduce vascular events, and (3) individualized treatment planning when cytoreduction is indicated in first- or second-line settings. Based on the randomized trial showing that roPEG-IFN is superior to HU in achieving durable hematologic and molecular remissions, guideline committees should reassess whether the drug merits preferred treatment status in the front-line setting.
Regardless of drug choice, management plans need to integrate baseline patient features and treatment considerations, such as comorbidities, symptom burden, drug toxicity profiles, and even public health concerns such as COVID-19 (Figure 2).91 Management of special situations such as major bleeding, anticoagulation options for thrombosis, surgery, and pregnancy is reviewed elsewhere.5,6,49 Aspirational goals such as delaying disease progression and extending survival should be part of the patient conversation but should acknowledge the paucity of data for these endpoints. This is particularly relevant to younger patients, in whom data have highlighted the underappreciated burden of PV on survival.92 The establishment of multi-institutional collaborations such as the MPN Research Consortium, development of MPN-focused Specialized Programs of Research Excellence, and creation of partnerships between academia and MPN patient advocacy groups will provide much-needed resources to narrow the assessment gap and tackle big vision research agendas.
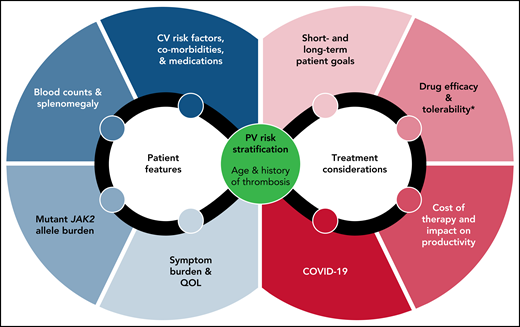
Considerations in the management of the patient with PV. Baseline PV risk, patient features, choice of cytoreduction, patient goals, and topical issues such as the COVID-19 pandemic need to be integrated in the treatment decision-making process. *Tolerability not only refers to drug-specific toxicity profiles but also therapy-related iatrogenesis of disease-related issues such as worsening of iron deficiency and hemorrhage. PV, polycythemia vera. QOL, quality of life.
Considerations in the management of the patient with PV. Baseline PV risk, patient features, choice of cytoreduction, patient goals, and topical issues such as the COVID-19 pandemic need to be integrated in the treatment decision-making process. *Tolerability not only refers to drug-specific toxicity profiles but also therapy-related iatrogenesis of disease-related issues such as worsening of iron deficiency and hemorrhage. PV, polycythemia vera. QOL, quality of life.
A recent analysis of IFN treatment of patients with MPNs suggested that achievement of a CHR for at least 24 months and a JAK2 V617F VAF <10% result in a lower cumulative incidence of post-discontinuation relapse compared with achievement of a CHR <24 months or JAK2 V617F VAF ≥10%.93 Therefore, treatment-free remission, which has been extensively studied in patients with chronic myeloid leukemia who are on tyrosine kinase inhibitor therapy,94 merits further study in IFN-treated patients with PV who obtain deep hematologic and molecular responses. Because the long runway of disease in younger individuals poses special challenges when contemplating whether to commit to prolonged therapy, a data-rich opportunity exists in these patients for evaluating the kinetics of treatment-free remission on roPEG-IFN.
For low-risk patients with PV, cytoreduction is recommended for several indications: new thrombosis or disease-related major bleeding, frequent and/or persistent need for phlebotomy, or poor tolerance of phlebotomy; (symptomatic) splenomegaly; progressive thrombocytosis and/or leukocytosis; and worsening disease-related symptoms.5,6 However, it is not clear whether low-risk patients with the rare need for phlebotomy (eg, 1-2 times annually) would benefit from further cytoreduction. The oft-asked clinical corollary is “How many phlebotomies in a year represent uncontrolled disease?”
In addition to the focus on new treatment strategies, future priorities in the treatment landscape of PV include further defining the role of IFNs in lower-risk disease and generating more biomarker data during intervention trials to better understand disease biology and predictors of response and progression.95,96 In the current era of precision medicine, the use of genome editing tools at the laboratory bench should help facilitate translational application of new theranostics for patients with PV.
Acknowledgments
The author is grateful to the Charles and Ann Johnson Foundation, Eli and Carmela Pasternak, Caroline Stokes, and members of the Stanford Hematology Clinical Research Group for their support of MPN/PV research. The author also acknowledges MPN colleagues for their generosity, guidance and patients for their participation in clinical trials to advance MPN research.
This perspective is dedicated to the memory of colleague Steven Coutré, whose leadership, mentorship, and kindness sustained his patients and the Stanford hematology family.
Authorship
Conflict-of-interest disclosure: J.G. received research funding from Incyte, Novartis, Kartos, Blueprint Medicines, Deciphera Pharmaceuticals, Cogent Biosciences, AbbVie, Celgene, BMS, and Protagonist Therapeutics. J.G. serves on advisory boards and receives honoraria for consulting from Incyte, Novartis, Kartos, Blueprint Medicines, Deciphera Pharmaceuticals, Cogent Biosciences, AbbVie, Celgene, BMS, Protagonist Therapeutics, and PharmaEssentia.
Correspondence: Jason Gotlib, Stanford Cancer Institute, 875 Blake Wilbur Dr, Room 2324, Stanford, CA 94305-6555; e-mail: jason.gotlib@stanford.edu.

