In this issue of Blood, 1 describe the identification and characterization of the novel cereblon-related E3 ligase modulator (CC-90009). Although cereblon-interacting agents (eg, immunomodulatory drugs; IMiDs) have established activity in multiple myeloma and myelodysplastic syndromes, these findings could have significant implications for the treatment of acute myelogenous leukemia (AML).

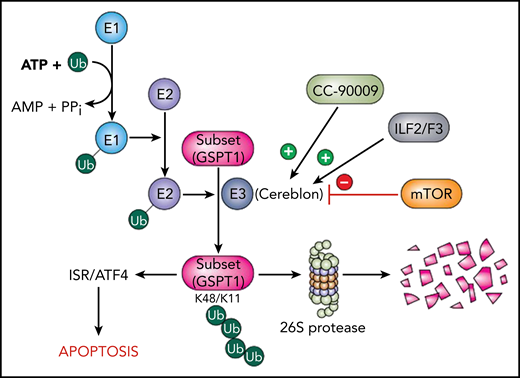
Schema of cereblon/CC-90009 interactions in the context of the UBS. Ubiquitin (Ub) is activated by a ubiquitin-activating enzyme (E1) and transferred to a ubiquitin-conjugating enzyme (E2) prior to linkage to protein substrates by a ubiquitin ligase (E3) complex, of which cereblon is a key component. The K48/K11 ubiquitination of substrates targets them for degradation by the 26S proteasome. CC-90009 promotes binding of cereblon to the translation termination protein GSPT1, which leads to the selective elimination of this protein. Loss of GSPT1 in AML cells triggers the ISR via ATF4, culminating in cell death. These events are opposed by loss of the alternative splicing modulators ILF2/ILF3, which diminish cereblon expression, as well as by hyperactivation of the mTOR pathway, which blocks GSPT1 degradation.
Schema of cereblon/CC-90009 interactions in the context of the UBS. Ubiquitin (Ub) is activated by a ubiquitin-activating enzyme (E1) and transferred to a ubiquitin-conjugating enzyme (E2) prior to linkage to protein substrates by a ubiquitin ligase (E3) complex, of which cereblon is a key component. The K48/K11 ubiquitination of substrates targets them for degradation by the 26S proteasome. CC-90009 promotes binding of cereblon to the translation termination protein GSPT1, which leads to the selective elimination of this protein. Loss of GSPT1 in AML cells triggers the ISR via ATF4, culminating in cell death. These events are opposed by loss of the alternative splicing modulators ILF2/ILF3, which diminish cereblon expression, as well as by hyperactivation of the mTOR pathway, which blocks GSPT1 degradation.
This group reported that CC-90009 coopts the cereblon (CBRN) ring ligase 4, leading to enhanced ubiquitination and subsequent degradation of the translation termination protein GSPT1, culminating in AML cell death. They further demonstrated that GSPT1 depletion plays a significant functional role in the anti-AML activity of CC-90009 and that this agent effectively eradicated AML stem cells in multiple AML patient-derived xenograft models. Mechanistically, the investigators used a genome-wide CRISPR-Cas9 screen to identify the ILF2/3 heterodimeric complex and the mTOR pathway as key regulators of CC-90009 activity. Finally, they showed that engagement of the ATF4-related integrated stress response (ISR) played a key functional role in antileukemic actions. Given that CC-90009 is currently undergoing phase 1 evaluation in AML (NCT02848001), such insights could have important clinical implications for AML therapy.
This study represents an extension of the investigators’ previous article in which they described the precursor CBRN modulator CC-885, which also targeted GSPT1, as well as multiple other substrates.2 The clinical development of this agent was hampered by multiple factors, including toxicity, presumably due to lack of specificity, as well as uncertainty regarding the mechanism(s) by which it triggered cell death. In contrast, CC-90009 is highly specific for GSPT1 degradation, raising the possibility that the absence of off-target toxicity could make this agent a superior clinical candidate compared with its predecessor.
CC-90009 is a member of an expanding class of antitumor agents exhibiting the ability to modulate the disposition of critical protein substrates by perturbing degradative pathways. Several of these agents converge upon ubiquitin E3 ligases that are responsible for K48/K11 ubiquitination of proteins, a process that targets them for degradation by the ubiquitin-proteasome system (UBS).3 However, modulation of the UBS can lead to enhanced degradation or accumulation of proteins, depending upon context. For example, proteasome inhibitors block the degradation of diverse proteins, including IκBα, leading to inactivation of the cytoprotective NF-κB pathway.4 Alternatively, proteolysis targeting chimeras (PROTACS) selectively eliminate oncogenic proteins by directly targeting them for E3 ubiquitin ligase–mediated degradation.5 NEDD8 inhibitors act by preventing neddylation and activation of E3 ligases, leading to accumulation of diverse proapoptotic proteins.6 Interest in CRBN as a therapeutic target was catalyzed by the observation that IMiDs (eg, lenalidomide) act, at least in part, in myeloma cells by modulating CRBN function to increase ubiquitination and degradation of important survival proteins (eg, the zinc-finger transcription factors IKFZ1/3).7 Moreover, the efficacy of IMiDs in del(5q) myelodysplastic syndrome has been attributed to CK1α depletion.8 However, the basis by which E3 ligase modulators might induce cell death in AML is essentially unknown.
Using a CRISPR-Cas screen, the investigators identified 3 new genetic pathways that influenced the ability of CC-90009 to disrupt cereblon-related events and, by extension, antileukemic activity. First, they found that modulators of RNA alternative splicing, such as ILF2/3, diminished cereblon expression and reduced CC-90009 activity. Second, they discovered that activation of the mTOR signaling pathway antagonized GSPT1 degradation and attenuated CC-90009–mediated cell killing. Finally, they demonstrated that CC-90009 exposure resulted in induction of various endoplasmic reticulum stress response elements (eg, ATF4) and that genetic ablation of these factors reduced CC-90009 efficacy. Importantly, they made the novel observation that GSPT1 degradation was necessary and sufficient to account for CC-90009 activity. Such findings provide a foundation for understanding the molecular basis for CC-90009 actions. A schematic diagram summarizing these observations and placing the CC-90009/cereblon/GSPT1 link within the context of the protein-degradative pathway is provided (see figure).
Collectively, these findings furnish a theoretical foundation for adding a cereblon modulator, such as CC-90009, to the therapeutic armamentarium for AML treatment, as well as suggest a number of possibilities for optimizing its use in the future. Furthermore, the mechanistic insights described in their article could help to identify biomarkers, allowing individualized CC-90009 treatment. For example, inasmuch as genetic ablation of interleukin-F2/F3 attenuated CC-90009 efficacy, it is conceivable that cells with high basal expression of these RNA alternative splicing genes might be particularly sensitive to this agent. Analogously, cells with high expression of GSPT1 may be addicted to this translation termination protein and, as a consequence, be particularly vulnerable to its loss following cereblon modulation–mediated degradation. Additionally, the observation that hyperactivation of the mTOR pathway attenuated CC-90009 activity by antagonizing GSPT1 degradation suggests that cells in which this pathway is intrinsically activated may display CC-90009 resistance. Answers to these questions await further preclinical investigation; results emerging from ongoing trials of CC-90009 in AML could be informative.
The present observations could also provide insights into rational combination therapies involving cereblon modulators, such as CC-90009, in AML and potentially other malignancies. For example, given that intrinsic activation of the mTOR pathway significantly reduced CC-90009 activity, it would be logical to explore the possibility that mTOR inhibitors, such as rapamycin,9 might synergize with CC-90009 and/or overcome resistance to this agent. Preclinical studies confirming or refuting this possibility could easily be conducted.
Finally, the present study, while providing significant information about the mechanism of action of CC-90009, leaves a number of questions to be addressed. For example, the mechanism by which GSPT1 depletion induces the ISR and triggers cell death in AML cells remains to be defined. In addition, it still needs to be determined whether an agent, such as CC-90009, that induces protein degradation by modulating E3 ligase activity and selectively reduces GSPT1 abundance will prove superior to PROTACs, which exhibit similar selectivity. Will the specificity of CC-90009 toward GSPT1 be associated with diminished toxicity in humans? What mechanisms are responsible for intrinsic or acquired resistance to this agent? Given the current degree of interest in modulators of protein degradation in AML and other hematopoietic malignancies, it is very likely that answers to these questions will be forthcoming shortly.
Conflict-of-interest disclosure: The author declares no competing financial interests.