Key Points
Ibrutinib can result in durable disease control in patients with HCL who are not expected to benefit from purine analogs.
The safety profile of ibrutinib in HCL is similar to that in other diseases with cytopenias and risk of frequent infections.

Abstract
Hairy cell leukemia (HCL) is a rare B-cell malignancy, and there is a need for novel treatments for patients who do not benefit from purine analogs. Ibrutinib, an oral agent targeting Bruton tyrosine kinase in the B-cell receptor signaling pathway, is highly effective in several malignancies. Its activity in HCL was unknown, so we conducted a multisite phase 2 study of oral ibrutinib in patients with either relapsed classic or variant hairy cell leukemia. The primary outcome measure was the overall response rate (ORR) at 32 weeks, and we also assessed response at 48 weeks and best response during treatment. Key secondary objectives were characterization of toxicity and determination of progression-free survival (PFS) and overall survival (OS). Thirty-seven patients were enrolled at 2 different doses (24 at 420 mg, 13 at 840 mg). The median duration of follow-up was 3.5 years (range, 0-5.9 years). The ORR at 32 weeks was 24%, which increased to 36% at 48 weeks. The best ORR was 54%. The estimated 36-month PFS was 73% and OS was 85%. The most frequent adverse events were diarrhea (59%), fatigue (54%), myalgia (54%), and nausea (51%). Hematologic adverse events were common: anemia (43%), thrombocytopenia (41%), and neutropenia (35%). Ibrutinib can be safely administered to patients with HCL with objective responses and results in prolonged disease control. Although the initial primary outcome objective of the study was not met, the observation of objective responses in heavily pretreated patients coupled with a favorable PFS suggests that ibrutinib may be beneficial in these patients. This trial was registered at www.clinicaltrials.gov as #NCT01841723.
Introduction
Hairy cell leukemia (HCL) is a rare chronic B-cell leukemia.1-3 The term HCL encompasses both the classic (cHCL) and variant (vHCL) forms, which are now recognized by the World Health Organization to be biologically distinct entities.4 cHCL carries the BRAF p.V600E mutation and has excellent long-term outcomes in the majority of patients, whereas vHCL behaves more aggressively and has shorter progression-free survival (PFS) and overall survival (OS) rates.5-7 Despite the development of the purine nucleoside analogs (PNAs) pentostatin and cladribine, which are highly effective in treating cHCL, the 5-year disease-free survival after treatment with these agents is only 77%, and 36% to 44% of patients relapse and require subsequent therapy.8-10 Patients who have a short duration of remission after treatment with PNAs or who are unable to receive them have limited treatment options. Furthermore, the use of repeated cycles of purine analogs for patients in relapse results in prolonged immunosuppression with delayed recovery of immune effector cells lasting months to longer than a year. These agents that produce profound and prolonged lymphopenia may result in decreased immunity to viruses and other dangerous pathogens.
Consequently, there is increased interest in using agents that are less immunosuppressive to achieve remission. BRAF inhibitors such as vemurafenib and the immunotoxin conjugate moxetumomab pasudotox are effective and less myelosuppressive or immunosuppressive, but the duration of remission varies with the quality of the response achieved.11,12 Patients who achieve a complete response (CR) with vemurafenib may remain in remission for more than a year, whereas those with a partial remission relapse earlier. Moxetumomab pasudotox produced a durable CR rate of 30%, with 33.8% of patients achieving CR with negative minimal residual disease (MRD). The median duration of complete remission was not reached for those with negative MRD, whereas the median duration of response for those with positive MRD was 5.9 months.13
Inhibition of B-cell receptor (BCR) signaling is an effective treatment strategy in several B-cell malignancies, including chronic lymphocytic leukemia (CLL), mantle cell lymphoma, marginal zone lymphoma, and lymphoplasmacytic lymphoma.14-18 These malignancies rely in part on constitutive BCR signaling for proliferation and survival. Ibrutinib, an orally bioavailable covalent inhibitor of Bruton tyrosine kinase (BTK), interrupts signaling through the BCR pathway and is approved for treatment of these cancers.19 Given the efficacy of ibrutinib in other B-cell malignancies, we hypothesized that it would be effective for HCL. We therefore performed a multicenter phase 2 study of ibrutinib in HCL patients with relapsed disease for whom PNAs were unsuitable, or in untreated patients with vHCL for whom there is no accepted standard treatment. This article describes the clinical benefit and associated toxicity of ibrutinib alone in patients with heavily pretreated relapsed HCL and also lays the groundwork for potential studies involving a strategic combination of active agents.
Methods
Patients
Patients at least 18 years of age with a diagnosis of HCL and an indication for therapy were enrolled.4,20 Patients with cHCL had to have previously received a PNA or be deemed medically unfit to receive one. Both previously treated and untreated patients with vHCL were eligible. Full eligibility criteria are listed in the supplemental Data, available on the Blood Web site.
Trial design and patient treatment
This study was a phase 2 multisite, open-label, single-agent study funded by the National Cancer Institute, and participating sites are listed in the supplemental Data. Ibrutinib is provided by the National Cancer Institute through an agreement with Pharmacyclics. All patients provided informed consent. The study was conducted according to the principles of the Declaration of Helsinki and the International Conference on Harmonization Guidelines for Good Clinical Practice. The protocol was approved by the institutional review boards of all participating sites.
The study has a 2-stage design. If the type I and type II error rates are each constrained to 10%, a 2-stage phase 2 Simon optimal study design requires a minimum of 13 and a maximum of 31 evaluable patients to test the hypothesis that the overall response rate (ORR) at 32 weeks would be at least 55% and would not be less than 30%. If, at the end of stage 1, there was no evidence that the 420-mg dose was sufficiently active, a higher 840-mg dose would be implemented.
Patients received ibrutinib by once-per-day oral administration. The first 13 patients received 420 mg orally once per day. Because there was a lack of objective responses, the dose was increased to 840 mg. However, because responses occurred after 32 weeks with the 420-mg dose and because there were concerns regarding toxicity at the higher dose, the protocol was amended to treat all newly enrolled patients at 420 mg. Those who initially received the 840-mg dose could be switched to the 420-mg dose. Ibrutinib was continued until toxicity became unacceptable or until disease progressed as long as the patient was deriving clinical benefit.
The study was open to accrual on April 1, 2013, and remains open. The investigators decided to publish the results before reaching the initial accrual goal, considering the long duration of study follow-up required to assess response and the potential benefit to patients of disseminating the study results. This agent was observed to produce objective responses with prolonged PFS in a heavily pretreated group of patients. The increased use of this commercially available agent off-study merited publication of our results to provide information regarding potential prolonged control of relapsed/refractory (R/R) disease and toxicities that might be encountered. Data as of 9 September 2019 were analyzed for publication.
End points and assessments
The primary end point was the ORR (CRs and partial responses [PRs]) at 32 weeks. There was an additional planned assessment at 48 weeks, and after that, response was assessed at the discretion of the investigator. Key secondary end points included characterization of the toxicity and tolerability of ibrutinib in HCL, PFS, and OS.
CR was defined as near normalization of blood counts with hemoglobin >11 g/dL, absolute neutrophil count (ANC) >1.5 × 103/μL, and platelet count >100 × 103/μL, along with absence of hairy cells on bone marrow core biopsy, bone marrow aspirate, and peripheral blood by morphology, plus resolution of splenomegaly and/or lymphadenopathy by physical examination. Immunohistochemistry (IHC) or flow cytometry was not considered when assessing CR. CR was determined by achieving the required hematologic parameters and assessing the bone marrow aspirate and biopsy for the presence of leukemic cells morphologically characteristic of HCL. MRD was also assessed in each patient and was determined in patients with CR. MRD negativity was defined as the absence of detectable leukemia cells in the blood and bone marrow by flow cytometry and by IHC examination using specific markers for HCL in the bone marrow. Both were assessed according to institutional protocols at the treating institution.
PR was defined as near normalization of blood counts with persistent morphologic evidence of HCL and at least 50% reduction in lymphadenopathy or splenomegaly on physical examination. Progressive disease (PD) was defined as worsening cytopenias or failure to achieve transfusion or growth factor independence by 12 weeks, increase in lymphadenopathy by 50% in any index node or new lymphadenopathy, increase in splenomegaly by physical examination or radiography, or an increase in the percentage of hairy cells in the bone marrow by morphologic analysis. Stable disease (SD) was defined as not meeting criteria for CR, PR, or PD. All responses were determined by the treating investigator and secondarily verified by 2 investigators (K.A.R. and M.R.G.). Adverse events (AEs) were captured and graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events. To ensure the safe conduct of the study, a monthly safety call was conducted with all participating institutions. The safety data were also reviewed regularly at The Ohio State University during the multidisciplinary phase 1/2 program meeting.
Pharmacokinetic and pharmacodynamic analysis
To determine the pharmacokinetics (PK) of ibrutinib in patients with HCL, plasma samples were collected and analyzed for both ibrutinib and the ibrutinib metabolite dihydrodiol ibrutinib using a validated liquid chromatography-tandem mass spectrometry assay.21 Additional pharmacodynamic studies examined leukemia mobilization into the peripheral blood by using a flow cytometry assay, changes in immunoglobulin (Ig) levels during treatment, expression of phospho-ERK (pERK) by IHC, and sequencing of BTK and PLCG2. Detailed methods are provided in the supplemental Data.
Statistical analysis
Summary statistics were calculated for patient demographics and clinical characteristics. The maximum grade for each type of AE was recorded for each patient in frequency tables. Response rates were calculated with 95% exact binomial confidence intervals (CIs) and compared between histologic subtypes using the Fisher’s exact test at each time point. PFS was determined from the start of treatment to disease progression or death (whichever occurred first) or were censored at the date of last follow-up for patients alive without events. OS was determined from the treatment start date to death as a result of any cause or were censored at the date of last follow-up. PFS and OS with 95% CIs were estimated by using the Kaplan-Meier method. PFS and OS were also evaluated by dose level and histologic subtypes using the log-rank test. Univariable analysis was performed to examine baseline characteristics associated with response using logistic regression models, and PFS was assessed by using Cox proportional hazards models. Odds ratios and hazard ratios with 95% CIs were estimated for response and PFS, respectively.
Kruskal-Wallis tests were used to assess the correlation between best response and PK parameters. Statistical significance for PK parameters, AEs, and disease characteristics were determined by using Mann-Whitney U tests. Cox proportional hazards models were used to explore the association between PK parameters and PFS. P values of <.05 were considered statistically significant. All analyses were conducted in SAS version 9.4 (SAS Institute, Cary, NC). Graphic analyses were performed using GraphPad Prism version 8 (GraphPad Software, San Diego, CA).
Results
Patients and treatments
A total of 37 patients were enrolled with 76% (28 of 37) having cHCL and 24% (9 of 37) having vHCL. Baseline patient characteristics and laboratory values are provided in Table 1. Testing for BRAF p.V600E was completed in 36 patients, and 20 of them had the mutation (supplemental Table 1). Most patients had a long disease course with a median of 9.8 years (range, 0.2-42.1 years) since diagnosis. The median number of previous therapies was 4 (range, 0-12 previous therapies), and all previously treated patients had received PNAs. Two patients with vHCL were previously untreated (supplemental Table 2).
Twenty-four patients started treatment at 420 mg once per day, and 13 patients started treatment at 840 mg once per day. There were no significant differences in baseline characteristics between patients treated at the 2 different doses except in absolute lymphocyte count (ALC) and subtype (Table 1). Most cycles (78%) were given at 420 mg (supplemental Table 3).
At a median follow-up of 3.5 years (range, 0-5.9 years) for all patients, 15 patients were still receiving study treatment, and 22 patients had discontinued treatment. Reasons for discontinuation were PD in 9 patients (41%), AEs in 7 (32%), patient or investigator decision in 4 (18%), and death during treatment in 2 (9%), both as a result of pneumonia (supplemental Table 4).
Efficacy
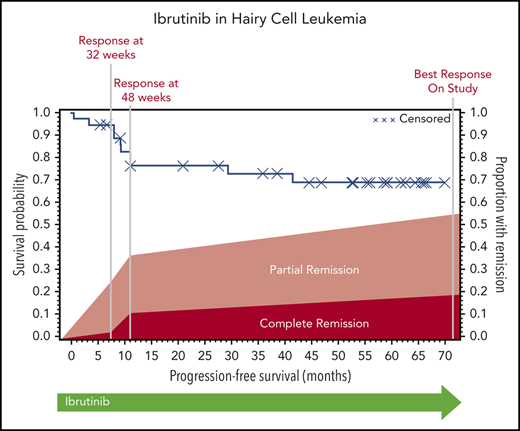
The ORR (CR and PR) at 32 weeks was 24% (95% CI, 12-41), and this improved to 36% (95% CI, 21-54) at 48 weeks. Response could be assessed at the investigator’s discretion after 48 weeks; the ORR could be assessed at any time after the start of ibrutinib treatment and was calculated as 54% (95% CI, 37-71). At 32 weeks, 1 patient had a CR, 8 had a PR, 21 had SD, 3 had PD, and 4 did not undergo assessment because they had discontinued treatment for lack of response (n = 3) or death (n = 1). The best response at any time on study was CR for 7 patients, PR for 13 patients, and SD for 10 patients. Of the patients with CR, 3 were MRD negative (supplemental Table 5). Responses are shown in Figure 1. Response rate was not significantly different between cHCL and vHCL subtypes, and ORR at any time was 54% and 56%, respectively (supplemental Table 6).
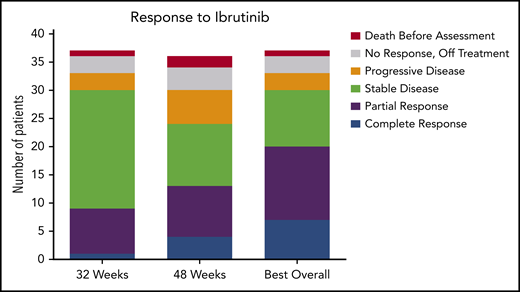
Response to ibrutinib. Response to ibrutinib is shown at both preplanned response assessments at 32 and 48 weeks. Patients with either a CR or a PR were considered responders. Patients who died before the 32-week response assessment and those who were off-study and therefore not assessed for response are shown. These data are also found in supplemental Table 5. One patient is still receiving treatment but has not reached the 48-week assessment and is included in the 32-week assessment and best response for any cycle totals.
Response to ibrutinib. Response to ibrutinib is shown at both preplanned response assessments at 32 and 48 weeks. Patients with either a CR or a PR were considered responders. Patients who died before the 32-week response assessment and those who were off-study and therefore not assessed for response are shown. These data are also found in supplemental Table 5. One patient is still receiving treatment but has not reached the 48-week assessment and is included in the 32-week assessment and best response for any cycle totals.
The median PFS was not reached, and the estimated 36-month PFS rate was 73%. The median OS was 69 months, and the estimated 36-month OS rate was 85% (Figure 2A). There were no differences in PFS or OS between histologic subtypes (P = .88; P = .76) or dose level (P = .24; P = .95, respectively) (supplemental Figures 1 and 2). Figure 2B shows individual patient outcomes. To determine baseline characteristics associated with response or PFS, we performed a univariable analysis that included sex, age, histologic subtype, BRAF mutation status, previous splenectomy, and previous treatments. We found that only age was associated with response; the odds ratio was 0.90 (95% CI, 0.84-0.98) per 1-year increase, which indicated lower likelihood of response with increasing age (supplemental Table 7). Age was not associated with risk for progression or death.
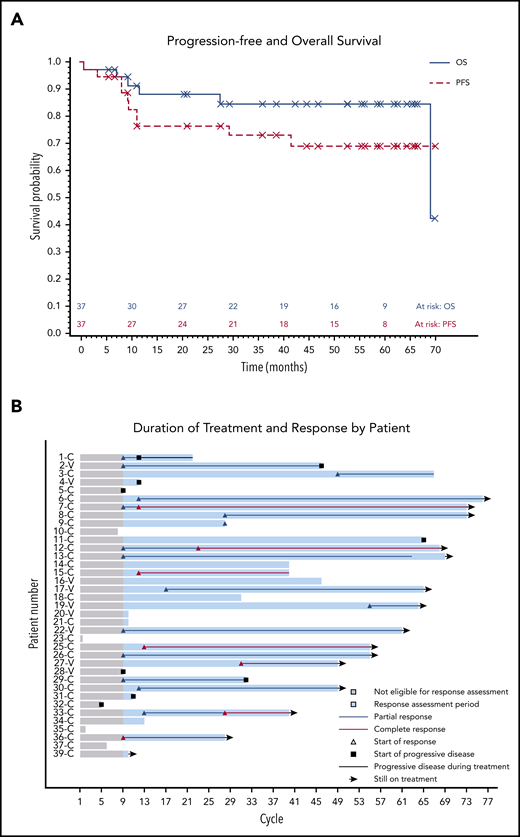
Patient survival and response. (A) PFS and OS after starting ibrutinib. The median for PFS was not reached, and the median OS was 69.1 months. (B) Swimmers plot with outcomes for each individual patient. Gray shading shows time before the first formal response assessment. Before that, patients were assessable for disease progression and survival but not remission. Patient 1 continued to receive treatment despite PD in the marrow because of the clinical benefit. Patients who discontinued therapy without disease progression discontinued for either AEs or death. Note that there is no patient 24 or patient 38 because those patients were found to be ineligible at screening and they received a study number but did not start study treatment. C, classic; V, variant.
Patient survival and response. (A) PFS and OS after starting ibrutinib. The median for PFS was not reached, and the median OS was 69.1 months. (B) Swimmers plot with outcomes for each individual patient. Gray shading shows time before the first formal response assessment. Before that, patients were assessable for disease progression and survival but not remission. Patient 1 continued to receive treatment despite PD in the marrow because of the clinical benefit. Patients who discontinued therapy without disease progression discontinued for either AEs or death. Note that there is no patient 24 or patient 38 because those patients were found to be ineligible at screening and they received a study number but did not start study treatment. C, classic; V, variant.
In patients with baseline cytopenias, the median time to an ANC >1 × 103/μL was 2 cycles (95% CI, 1.8-5 cycles), platelet count >100 × 103/µL was 5 cycles (95% CI, 2-11 cycles), and hemoglobin >11 g/dL was 5 cycles (95% CI, 1.3-7 cycles) (supplemental Figure 3). The change in peripheral counts over the course of treatment is shown in supplemental Figure 4; most patients who were still being treated beyond cycle 12 had improvement in ANC, hemoglobin, and platelet count compared with baseline.
Safety
Hematologic AEs attributed to study treatment were common, with 43% of patients experiencing anemia (5% grade ≥3), 41% thrombocytopenia (22% grade ≥3), and 35% neutropenia (22% grade ≥3) (Table 2). Despite 13 patients experiencing neutropenia, there were only 4 who had febrile neutropenia.
Common nonhematologic AEs attributed to study treatment were diarrhea (59%), fatigue (54%), myalgias (54%), nausea (51%), upper respiratory tract infection (46%), and bruising (43%). Cardiovascular AEs are frequently seen with ibrutinib in other diseases and were also seen in this study: hypertension (43%; 11% grade ≥3), atrial fibrillation (16%; none grade ≥3), palpitations (16%; none grade ≥3), sinus bradycardia (8%; none grade ≥3), atrial flutter (5%; 3% grade ≥3), and heart failure (3%; all grade ≥3). All treatment-related and all-cause AEs are listed in supplemental Tables 8 and 9. When all AEs occurring in ≥20% of patients or grade ≥3 were examined, there were no significant differences in frequency between the 420-mg and 840-mg dose groups (supplemental Table 10).
Seven patients discontinued treatment because of AEs such as decreased ejection fraction, palpitations, hypersensitivity to ibrutinib, thrombocytopenia, neutropenia, persistent cytopenias, and colon cancer (supplemental Table 4). There were 5 deaths during the study: 2 occurred during ibrutinib treatment, both as a result of pneumonia. Three deaths occurred during postibrutinib follow-up: 2 were the result of PD, and 1 was the result of a fungal infection after starting a subsequent treatment of HCL.
PK and pharmacodynamic measures
PK measurements were used to determine ibrutinib exposure. Because of the lack of circulating disease, BTK occupancy assays and other measures could not be reliably performed. PK parameters were estimated in 20 patients for day 1 with sampling out to 24 hours. Area under the curve and maximum concentration (Cmax) were not significantly higher in the 840-mg dose group compared with the 420-mg dose group (nonparametric Student t test, P > .05). Noncompartmental PK parameters were estimated (supplemental Table 11), and plasma concentration-time plots were generated (supplemental Figure 5). Ibrutinib PK in this patient population was similar to that reported in other populations.22
There was no significant association between ibrutinib PK and best response or PFS (supplemental Figure 6; supplemental Table 12). PK parameters were unaltered by disease characteristics, including histologic subtype, BRAF p.V600E mutation status, and previous splenectomy (supplemental Figure 7). There were no notable correlations with any frequent or severe AEs (supplemental Figure 8).
To determine whether ibrutinib causes mobilization of leukemia into the peripheral blood, the absolute numbers of CD19+ B cells in the blood were measured during the first 4 weeks of treatment. There were no patients with a substantial increase in circulating leukemia cells (supplemental Figure 9). In examining change in ALC over the course of treatment, 2 patients had an increase in ALC (supplemental Figure 4).
Because ibrutinib inhibits BTK, which is essential to the functioning of healthy B cells, Ig levels were measured out to 18 months of treatment. There was a significant decrease in IgG from baseline with no change in IgA or IgM (supplemental Figure 9).
The presence of pERK was assessed because it is a prosurvival signal in HCL and is downstream of BTK in BCR signaling.23,24 Available bone marrows were examined for pERK using IHC (Figure 3A). Notably, several patients with persistent presence of pERK after 32 and 48 weeks of treatment continued to have a durable benefit from ibrutinib (Figure 3B).
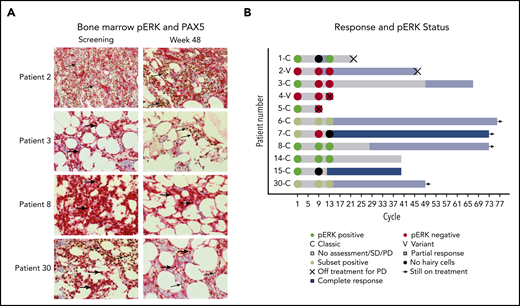
pERK status during treatment and clinical outcome. (A) pERK expression in HCL cells in the bone marrow are shown for 4 representative patients at screening and week 48 of ibrutinib treatment. pERK expression is shown as red cytoplasmic staining. Leukemia cells were identified by a PAX5 stain with brown nuclear staining when present. The use of both stains identifies pERK expression specifically in the leukemia cells. The presence of cytoplasmic pERK is shown with thick arrows and the absence is shown with thin arrows. Patient 2 has vHCL and had a diffuse infiltrate of leukemic cells at both screening and week 48 of treatment. The leukemia cells were negative for pERK staining at every time point tested. Patients with vHCL have previously been shown to not have pERK, and this may not be a prosurvival signal in these patients, given that vHCL and cHCL have relatively different biology. The remaining 3 patients (3, 8, and 30) all have cHCL and a durable benefit from ibrutinib with a PFS of more than 2 years, and all were continuing to receive ibrutinib at the last follow-up. They all had different changes in pERK status after starting treatment. Patient 3 had pERK in the leukemia cells at screening that was absent by week 48, patient 8 had persistent presence of pERK at all time points tested, and patient 30 had a subset of pERK-positive leukemia cells at screening and at week 48. (B) Swimmer’s plot with pERK status at screening, week 32, and week 48 for all patients for whom samples were available. Both patients with vHCL (patient 2 and patient 4) were pERK negative at every time point tested, which is consistent with what was previously described in patients with vHCL. Many patients with persistence of pERK continued to receive ibrutinib without progression for an extended period. This supports that (contrary to what was shown with vemurafenib) the presence of pERK during ibrutinib treatment may not predict a shorter PFS and argues that the mechanism of effect of ibrutinib may not involve decreasing ERK phosphorylation.
pERK status during treatment and clinical outcome. (A) pERK expression in HCL cells in the bone marrow are shown for 4 representative patients at screening and week 48 of ibrutinib treatment. pERK expression is shown as red cytoplasmic staining. Leukemia cells were identified by a PAX5 stain with brown nuclear staining when present. The use of both stains identifies pERK expression specifically in the leukemia cells. The presence of cytoplasmic pERK is shown with thick arrows and the absence is shown with thin arrows. Patient 2 has vHCL and had a diffuse infiltrate of leukemic cells at both screening and week 48 of treatment. The leukemia cells were negative for pERK staining at every time point tested. Patients with vHCL have previously been shown to not have pERK, and this may not be a prosurvival signal in these patients, given that vHCL and cHCL have relatively different biology. The remaining 3 patients (3, 8, and 30) all have cHCL and a durable benefit from ibrutinib with a PFS of more than 2 years, and all were continuing to receive ibrutinib at the last follow-up. They all had different changes in pERK status after starting treatment. Patient 3 had pERK in the leukemia cells at screening that was absent by week 48, patient 8 had persistent presence of pERK at all time points tested, and patient 30 had a subset of pERK-positive leukemia cells at screening and at week 48. (B) Swimmer’s plot with pERK status at screening, week 32, and week 48 for all patients for whom samples were available. Both patients with vHCL (patient 2 and patient 4) were pERK negative at every time point tested, which is consistent with what was previously described in patients with vHCL. Many patients with persistence of pERK continued to receive ibrutinib without progression for an extended period. This supports that (contrary to what was shown with vemurafenib) the presence of pERK during ibrutinib treatment may not predict a shorter PFS and argues that the mechanism of effect of ibrutinib may not involve decreasing ERK phosphorylation.
We used sequencing of BTK and PLCG2 in 4 patients (2 cHCL, 2 vHCL) who had available samples from the time of developing PD as an exploratory analysis to ascertain mechanisms of resistance to ibrutinib in HCL. Mutations in BTK at the drug binding target (codon 481) or multiple regions of PLCG2, which is an immediate downstream target of BTK phosphorylation, are well described ibrutinib resistance mechanisms in CLL.25 No mutations in either BTK or PLCG2 were detected, which indicates that resistance mechanisms in HCL may be different.
Discussion
We found that in patients with heavily pretreated HCL, ibrutinib produced both responses and durable progression-free disease intervals. An ORR of 54% seems modest relative to that for PNAs; however, in our heavily pretreated population and in patients with the variant form of HCL, this result represents a definite benefit. This study did not meet the prespecified ORR of 50% at 32 weeks for the primary end point, but it did meet this goal if best overall response at any time was assessed. In addition, the long duration of disease control with ibrutinib in patients with extensive previous therapy for leukemia should not be overlooked.
In recent years, several novel agents have become available for patients in whom PNAs are unsuitable, but these drugs have both benefits and limitations. Vemurafenib, an inhibitor of BRAF p.V600E, has a high ORR of 96% to 100% (35% to 42% CRs) in cHCL with the BRAF p.V600E mutation. It has the advantage of being a time-limited treatment, but a limitation of vemurafenib is that the relapse-free survival can be short, with a median of 19 months in patients with a CR and only 6 months in those with a PR.11 In contrast to vemurafenib, ibrutinib results in durable disease control and is appropriate for treating patients without the BRAF p.V600E mutation.
Moxetumomab pasudotox, an immunotoxin drug conjugate targeting CD22, was recently approved by the US Food and Drug Administration for relapsed HCL. It has a high ORR (75%), but it does not have a toxicity profile acceptable to all patients with grade 3 or 4 hemolytic uremic syndrome (5%) or capillary leak syndrome (2.5%).13 Ibrutinib can cause toxicity, but its safety profile is different and may be acceptable for a different subset of patients. Many patients do not achieve MRD-negative status with moxetumomab pasudotox, and this is associated with a shorter PFS of a median 5.9 months.13
In retrospect, the primary end point assessment at 32 weeks was too early to observe the full effects of ibrutinib.26 Similar to responses in CLL, most responses were partial, and deeper responses occurred with longer treatment duration. The response criteria in this study were designed for cytotoxic agents and require recovery of hemoglobin, platelets, and neutrophil count for even a PR.20,27 Therefore, patients with a clinical response but continued mild ibrutinib-related cytopenias will have SD and long PFS, as was seen for a substantial number of patients.
The AEs of ibrutinib in HCL were like those established in other diseases with common AEs such as diarrhea, myalgia, fatigue, nausea, and bruising.15 The frequency of cardiovascular AEs such as atrial fibrillation and hypertension were similar as well. This suggests that the larger experience of using ibrutinib in these other diseases may help clinicians select patients with HCL who would be appropriate to receive this agent and to manage ibrutinib toxicities in this HCL population. Two patients died early during treatment as a result of infection, which is a common cause of death in patients with HCL and may not be the result of ibrutinib alone.28
In this study, there was no dose-related profile of toxicity. And there were no apparent meaningful associations between PK parameters and disease control as measured by both best response and PFS or AEs. Because the overall response and toxicity data were not statistically different between the 2 dose levels examined, it seemed prudent to complete the treatment with the 420-mg dose of ibrutinib.
Similar to what is seen in CLL, serum IgG significantly decreased from baseline with ibrutinib treatment.29 It is not clear what implications this has for risk of infection in HCL because the immune environment is different from that in CLL.
The mechanism of the effect of ibrutinib in HCL is not entirely clear, and there are several differences from the CLL experience. Distinct from what is seen in CLL, there was no mobilization of leukemia cells into the peripheral blood in the majority of patients.30 Interestingly, persistence of pERK in leukemia cells did not seem to result in a shorter PFS as has been shown with vemurafenib in HCL, and the absence of pERK cannot be used as a biomarker of response.11 ERK phosphorylation is decreased by ibrutinib in CLL because BCR signaling is interrupted, but this was not seen in HCL. This argues that the mechanism of action of ibrutinib may not be solely to decrease ERK phosphorylation and that ibrutinib may have other effects on leukemia cells or the immune environment.23 Furthermore, the mechanisms of resistance to ibrutinib are not the same as those in CLL. Among the 4 samples analyzed for BTK and PLCG2 mutations, none had CLL/B-cell lymphoma-type resistance-associated mutations, indicating that these are unlikely to be as frequent as in ibrutinib-resistant CLL. More work is needed to determine the mechanisms of effect and resistance to ibrutinib in HCL.
It is worth noting that the group of patients with HCL treated in this study was unusual. The majority had experienced a high number of treatments during their disease course and most did not derive prolonged benefit from purine analogs. A substantial fraction of patients with classic histology were noted to be negative for the BRAF p.V600E mutation. This may be a result of differences in testing, which was not performed centrally or was performed by sequencing on samples in which the number of leukemia cells might have been low, thus limiting the ability to detect the mutation, or it might be related to the unusual biology of patients in this cohort. Specifically, patients with an immunophenotypic profile of cHCL who were negative for a BRAF mutation have been found to have a distinct molecular profile and may harbor a mutation in MAP2K1 and/or IGVH4-34 B-cell receptor. These cHCL patients often do not respond to standard purine analogs, and they follow a difficult course with multiple relapses, consistent with the population recruited in this study.31 The intentional recruitment of patients with R/R may explain the high frequency of patients with cHCL who were negative for BRAF p.V600E. Although it was not performed in this study, extensive genotypic investigation would be of interest in this unusual patient population characterized as having cHCL with a negative BRAF p.V600E mutation.
Ibrutinib also showed an important benefit for patients with vHCL. The study was not designed to detect differences in outcome between cHCL and vHCL, and a post hoc analysis did not show a significant difference in ORR, PFS, or OS. The total of 9 patients reported in this study represents a relatively large group of patients with vHCL. Because vHCL generally has worse disease outcomes with treatments such as PNAs and a shorter survival than cHCL, this is an important finding. The disease control with ibrutinib may be more meaningful in this group of patients with fewer effective treatment options.6,7 This study included 2 patients with vHCL who were previously untreated, and ibrutinib may be appropriate initial therapy for some patients with vHCL.
In summary, ibrutinib has substantial therapeutic benefit with durable disease control in patients with HCL that have R/R disease or with the variant histology. The safety profile of ibrutinib in HCL is like that in other diseases and is appropriate for patients in whom chemotherapy may not be well tolerated. Given the efficacy of PNAs, these agents should continue to be used as an initial treatment in patients who can tolerate them.32-34 Ibrutinib represents an important new option resulting in durable disease control for those patients with HCL who cannot tolerate or are not expected to benefit from other available therapies. The results of this trial support the potential for further studies of the combination of ibrutinib with other active agents, a strategy that has been highly successful in other B-cell malignancies.
Presented at the Annual Meeting of the European Hematology Association, Barcelona, Spain, 18-20 September 2015, and the 58th Annual Meeting and Exposition of the American Society of Hematology, San Diego, CA, 3-6 December 2016.
Publication-related data are available by sending an e-mail to Kerry A. Rogers at kerry.rogers@osumc.edu.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors acknowledge members of The Ohio State University Multisite Clinical Trials Program, including Jennifer Sexton, Alison Neal, Rebecca Imboden, and Rana Roberts; members of The Ohio State University Clinical Trials Office, including Terri Hutchinson, for supporting this study; and Theresa Yu, Lieutenant, United States Public Health Service/National Cancer Institute for her contribution to this study.
This work was supported, in part, by The Ohio State University Comprehensive Cancer Center, by grants from the National Institutes of Health, National Cancer Institute (UM1CA186712 and P30 CA016058), and by the Hairy Cell Leukemia Foundation. K.A.R. is a Scholar in Clinical Research of the Leukemia & Lymphoma Society (CDP 2331-20).
Authorship
Contribution: J.A.J., M.R.G., and S.P.I. designed and implemented the study; K.A.R., L.A.A., J.S.B., D.C., T.C., L.R.J., R.J.K., F.R., C.A.S., W.E.C., J.A.J., and M.R.G. performed the study and treated study participants; L.W., E.M.M., and A.S.R. designed and performed the statistical analysis and created the figures; M.P. and A.D. analyzed the pharmacokinetic data; L.G., G.L., M.A., and D.M.L. designed and performed correlative studies; D.J. and A.N. performed sequencing; K.A.R., L.A.A., and M.R.G. wrote the first draft of the manuscript; and all authors had access to the data, analyzed and interpreted the data, reviewed the manuscript, and agreed with submission for publication.
Conflict-of-interest disclosure: K.A.R. receives research funding from AbbVie, Genentech, and Janssen, and participated in advisory boards for Acerta Pharma, Pharmacyclics, AbbVie, Genentech, Innate Pharma, and AstraZeneca. L.A.A. consults for AstraZeneca. A.S.R. serves on an independent data safety monitoring board for Telios Pharma. M.A. has consulted for AstraZeneca. J.S.B. has received consulting fees from AbbVie, AstraZeneca, and KITE Pharma. A.D. is employed by Gilead. R.J.K. receives research support from agreements between the National Institutes of Health and AstraZeneca, Genentech, Teva, and Novartis and is a coinventor on the National Institutes of Health patent for moxetumomab pasudotox. G.L. receives research funding from Genentech, Stemline, Novartis, and Beckman Coulter. D.J. receives research or testing support from Pharmacyclics, AbbVie, Novartis, MingSight, Acerta Pharma, ArQule, and Sunesis. C.A.S. served on a data safety monitoring board for Pharmacyclics and receives research funding for clinical trials. J.A.J. is employed by and has equity in Bristol Myers Squibb. M.R.G. has severed on a data safety monitoring committee for Acerta Pharma and Axio and has consulted for AstraZeneca and Pharmacyclics. The remaining authors declare no competing financial interests.
Correspondence: Kerry A. Rogers, Division of Hematology, The Ohio State University, 410 W. 12th Ave, Room 458, Columbus, OH 43210; e-mail: kerry.rogers@osumc.edu.