Key Points
Venetoclax and inotuzumab activate differential and synergistic apoptosis signals in B-lineage ALL cells.
Combination therapy may induce long-term treatment-free survival in B-lineage ALL-PDX models.

Abstract
Adult patients with relapsed B-cell precursor acute lymphoblastic leukemia (BCP-ALL) have a dismal prognosis. To improve pharmacotherapy, we analyzed induction of apoptosis by venetoclax and inotuzumab ozogamicin in terms of cytotoxicity and mode of action. Flow cytometry–based analyses of mitochondrial outer membrane permeabilization (MOMP) and ataxia telangiectasia mutated activation demonstrate rapid induction of MOMP by venetoclax and DNA damage signaling by inotuzumab ozogamicin, respectively. In primary ALL samples and patient-derived xenograft (PDX) models, venetoclax and inotuzumab ozogamicin cooperated and synergized in combination with dexamethasone in vitro in all tested samples of ALL. In murine PDX models, inotuzumab ozogamicin, but not venetoclax, induced complete remission in a dose-dependent manner but constantly failed to achieve relapse-free survival. In contrast, combination therapy with venetoclax, dexamethasone, and inotuzumab ozogamicin induced long-term leukemia-free survival and treatment-free survival in all 3 ALL-PDX models tested. These data demonstrate synergistic and highly efficient pharmacotherapy in preclinical models that qualify for evaluation in clinical trials.
Introduction
Adult patients with relapsed acute lymphoblastic leukemia (ALL) have a dismal prognosis with cure achievable only by allogeneic stem cell transplantation or chimeric antigen receptor T-cell therapy.1-4 Inotuzumab ozogamicin, which has been approved by the US Food and Drug Administration for treating adults with relapsed or refractory B-cell precursor ALL (BCP-ALL), couples the DNA double-strand break (DSB) inducer calicheamicin5 to an anti-CD22 antibody. Despite impressive complete remission (CR) rates and even minimal residual disease negativity, almost all patients in the INO-VATE ALL trial eventually relapsed without stem cell transplantation, demonstrating that inotuzumab ozogamicin monotherapy did not eradicate leukemia.2
We recently demonstrated rapid induction of apoptosis in BCP-ALL by BH3 mimetics such as venetoclax.6,7 Venetoclax directly releases apoptosis activators (mainly BIM) from BCL27 linked to permeabilization of the mitochondrial outer membrane (MOM), the point of no return for mitochondrial apoptosis.8 Venetoclax synergizes with other MOM permeabilization (MOMP)-sensitizing drugs such as dexamethasone and dasatinib in BCR-ABL+ ALL7,9,10 and proved curative in a xenograft BCR-ABL+ ALL mouse model.7
Study design
Primary samples were collected from patients who were newly diagnosed or had relapsed B-lineage ALL (see supplemental Table 1, available on the Blood Web site) who provided informed consent in accordance with the Declaration of Helsinki and the Ethics Committee of Hannover Medical School. All experimental procedures are described in the supplemental Materials.
Results and discussion
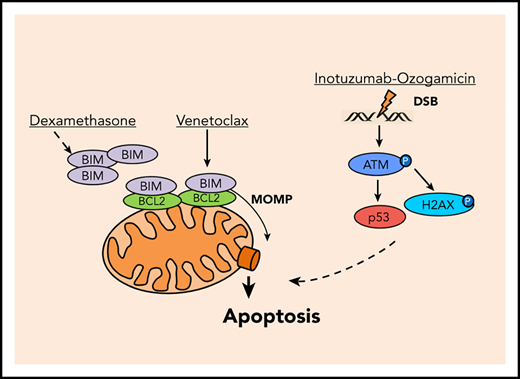
We here hypothesized that induction of MOMP by venetoclax and DSB apoptosis signaling by calicheamicin may cooperate in BCP-ALL (Figure 1A). Induction of MOMP is linked to a decay of the mitochondrial membrane potential directly measureable in viable cells upon mitochondrial staining with tetramethylrhodamine ethyl ester (TMRE)7 (supplemental Figure 1A; supplemental Methods). Similarly, DSB-induced phosphorylation of the serine/threonine kinase ataxia telangiectasia mutated (ATM) on serine 1981 (pATM) and its target histone H2AX on serine 139 (γH2AX) (for review, see Blackford and Jackson11 ) can be measured by flow cytometry12 (supplemental Figure 1B; supplemental Methods). We used BCP-ALL patient-derived xenograft (PDX) models (supplemental Table 1) and cell lines to analyze the induction of MOMP and the accumulation of pATM and γH2AX by venetoclax and inotuzumab ozogamicin, respectively. After 3 hours, venetoclax, but not inotuzumab ozogamicin or dexamethasone, decreased TMRE fluorescence (Figure 1B; supplemental Figure 1C). In contrast, only inotuzumab ozogamicin substantially induced pATM and γH2AX after 6 hours (Figure 1C-D; supplemental Figure 1D-E). No propidium iodide staining was detectable, but substantial caspase-3 cleavage was detectable in up to 15% of venetoclax-treated cells after 6 hours, which reflects the differential kinetics of activation (not execution) of apoptotic signaling by mitochondrial and pATM and γH2AX staining, respectively (supplemental Figure 1F-I). Dexamethasone did not directly induce MOMP or pATM or γH2AX at these early time points (Figure 1B-D; supplemental Figure 1C-E).
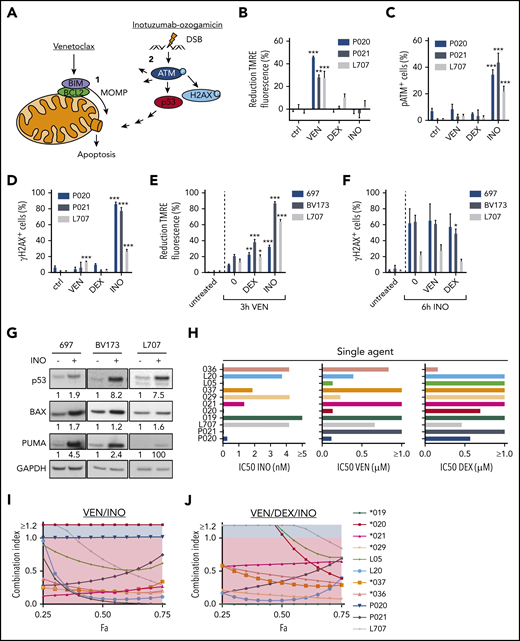
Induction of apoptosis in ALL cells by venetoclax (VEN), dexamethasone (DEX), and inotuzumab ozogamicin (INO). (A) Model of apoptosis induction by venetoclax and inotuzumab ozogamicin. Venetoclax directly induces MOMP by interrupting protein-protein interactions of anti- and proapoptotic proteins BCL2 and BIM. Release of BIM results in pore formation and subsequent apoptosis. Inotuzumab ozogamicin induces DSBs, which leads to phosphorylation of ATM kinase. ATM in turn phosphorylates H2AX and activates p53 signaling. (B-D) Mode of action of venetoclax and inotuzumab ozogamicin. P020, P021, and L707 PDX cells were treated with venetoclax (P020, 50 nM; P021, 1000 nM; L707, 250 nM), dexamethasone (P020, 1 µM; P021, 5 µM; L707, 1 µM), or inotuzumab ozogamicin (P020, 100 ng/mL; P021, 100 ng/mL; L707, 1000 ng/mL), respectively. Error bars indicate ± standard deviation (n = 3). (B) After 3 hours of treatment, cells were stained with tetramethylrhodamine ethyl ester (TMRE) to determine MOMP of viable cells by flow cytometry. Treatment with the carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP) ionophore served as positive control. Reduction of TMRE fluorescence of untreated cells was set as 0%. After 6 hours of treatment, cells were fixed, permeabilized, and stained with an antibody to (C) pATM or (D) γH2AX. (E) MOMP induction by venetoclax with or without pretreatment for 24 hours with dexamethasone (L707, 2 µM; 697, 5 µM; BV173, 200 nM) or inotuzumab ozogamicin (L707, 500 ng/mL; 697, 20 ng/mL; BV173, 20 ng/mL). After 3 hours of treatment, cells were stained with 50 nM TMRE to determine MOMP induction by flow cytometry. FCCP served as a positive control. Reduction of TMRE fluorescence of untreated cells was set as 0%. (F) γH2AX induction by inotuzumab ozogamicin in L707, 697, and BV173 cells with or without pretreatment for 24 hours with dexamethasone (L707, 5 µM; 697, 5 µM; BV173, 1 µM) or venetoclax (L707, 100 nM; 697, 250 nM; BV173, 250 nM). After 6 hours of treatment with inotuzumab ozogamicin (L707, 500 ng/mL; 697 and BV173, 100 ng/mL), cells were fixed, permeabilized, and stained with an antibody to γH2AX. (G) Western blot analysis for p53, BAX, and PUMA expression of L707, 697, and BV173 cells treated with 20, 2.5, or 1 ng/mL inotuzumab ozogamicin for 24 hours, respectively. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) served as loading control. Western blots are representative of 3 independent experiments. (H-J) Primary ALL samples and PDX cells were treated with inotuzumab ozogamicin, venetoclax, and dexamethasone alone or in combination in fixed ratios for 48 hours followed by CD19 antibody and propidium iodide staining. (H) IC50’s of inotuzumab ozogamicin, venetoclax, and dexamethasone were determined by CompuSyn software. (I-J) Fraction affected (Fa)/CI plots for drug combinations of venetoclax-inotuzumab ozogamicin (I) and venetoclax-dexamethasone-inotuzumab ozogamicin (J). CIs were calculated using CompuSyn software. CI <1 was considered synergistic. P values were calculated by 1-way analysis of variance (ANOVA) with Bonferroni correction. *P < .05; **P < .01; ***P < .001. ctrl, control.
Induction of apoptosis in ALL cells by venetoclax (VEN), dexamethasone (DEX), and inotuzumab ozogamicin (INO). (A) Model of apoptosis induction by venetoclax and inotuzumab ozogamicin. Venetoclax directly induces MOMP by interrupting protein-protein interactions of anti- and proapoptotic proteins BCL2 and BIM. Release of BIM results in pore formation and subsequent apoptosis. Inotuzumab ozogamicin induces DSBs, which leads to phosphorylation of ATM kinase. ATM in turn phosphorylates H2AX and activates p53 signaling. (B-D) Mode of action of venetoclax and inotuzumab ozogamicin. P020, P021, and L707 PDX cells were treated with venetoclax (P020, 50 nM; P021, 1000 nM; L707, 250 nM), dexamethasone (P020, 1 µM; P021, 5 µM; L707, 1 µM), or inotuzumab ozogamicin (P020, 100 ng/mL; P021, 100 ng/mL; L707, 1000 ng/mL), respectively. Error bars indicate ± standard deviation (n = 3). (B) After 3 hours of treatment, cells were stained with tetramethylrhodamine ethyl ester (TMRE) to determine MOMP of viable cells by flow cytometry. Treatment with the carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP) ionophore served as positive control. Reduction of TMRE fluorescence of untreated cells was set as 0%. After 6 hours of treatment, cells were fixed, permeabilized, and stained with an antibody to (C) pATM or (D) γH2AX. (E) MOMP induction by venetoclax with or without pretreatment for 24 hours with dexamethasone (L707, 2 µM; 697, 5 µM; BV173, 200 nM) or inotuzumab ozogamicin (L707, 500 ng/mL; 697, 20 ng/mL; BV173, 20 ng/mL). After 3 hours of treatment, cells were stained with 50 nM TMRE to determine MOMP induction by flow cytometry. FCCP served as a positive control. Reduction of TMRE fluorescence of untreated cells was set as 0%. (F) γH2AX induction by inotuzumab ozogamicin in L707, 697, and BV173 cells with or without pretreatment for 24 hours with dexamethasone (L707, 5 µM; 697, 5 µM; BV173, 1 µM) or venetoclax (L707, 100 nM; 697, 250 nM; BV173, 250 nM). After 6 hours of treatment with inotuzumab ozogamicin (L707, 500 ng/mL; 697 and BV173, 100 ng/mL), cells were fixed, permeabilized, and stained with an antibody to γH2AX. (G) Western blot analysis for p53, BAX, and PUMA expression of L707, 697, and BV173 cells treated with 20, 2.5, or 1 ng/mL inotuzumab ozogamicin for 24 hours, respectively. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) served as loading control. Western blots are representative of 3 independent experiments. (H-J) Primary ALL samples and PDX cells were treated with inotuzumab ozogamicin, venetoclax, and dexamethasone alone or in combination in fixed ratios for 48 hours followed by CD19 antibody and propidium iodide staining. (H) IC50’s of inotuzumab ozogamicin, venetoclax, and dexamethasone were determined by CompuSyn software. (I-J) Fraction affected (Fa)/CI plots for drug combinations of venetoclax-inotuzumab ozogamicin (I) and venetoclax-dexamethasone-inotuzumab ozogamicin (J). CIs were calculated using CompuSyn software. CI <1 was considered synergistic. P values were calculated by 1-way analysis of variance (ANOVA) with Bonferroni correction. *P < .05; **P < .01; ***P < .001. ctrl, control.
Next, sequential treatment was used to analyze interactions between direct induction of MOMP and DSB signaling (Figure 1E-F) in L707 PDX cells expandable on mesenchymal stem cells13,14 and in 697 and BV173 cells. Cells were preincubated with venetoclax, dexamethasone, and inotuzumab ozogamicin for 24 hours; for the last 3 hours, venetoclax was added, and for the last 6 hours, inotuzumab ozogamicin was added. Venetoclax induced MOMP compared with untreated controls. Preincubation with inotuzumab ozogamicin and dexamethasone further enhanced this process (Figure 1E) and increased relative BIM loading to BCL2 (supplemental Figure 1J). In contrast, inotuzumab ozogamicin induced γH2AX expression compared with untreated controls, but γH2AX levels remained unchanged after preincubation with venetoclax or dexamethasone, which suggests that these drugs may not sensitize cells for DSB responses (Figure 1F). Immunoblot analyses demonstrated that inotuzumab ozogamicin induced expression of p53 and its targets BAX and PUMA (Figure 1G). These data are in line with directly induced MOMP apoptosis or DSB signaling at least partly converging on components of the mitochondrial apoptosis machinery.
We next analyzed venetoclax, inotuzumab ozogamicin, and dexamethasone alone and in fixed ratios15 in primary ALL and PDX samples in coculture with mesenchymal stem cells (supplemental Table 1).7,14 Cell death was assessed by propidium iodide and human CD19 (huCD19) staining via flow cytometry after 48 hours. Drug responses were heterogeneous among ALL samples, and inotuzumab ozogamicin was effective in all but 1 sample (50% inhibitory concentration [IC50] <10 nM Figure 1H). Venetoclax was effective in 7 of 11 samples, and dexamethasone was effective in 4 of 11 samples (IC50 <1 µM). Next, combination therapies were analyzed using Chou-Talalay combination indices (CIs) with CI <1 indicative of drug synergy.15 The combination of venetoclax and inotuzumab ozogamicin reduced drug IC50 values (supplemental Figure 1K) and behaved in a highly synergistic and additive manner (CI, 1; P020 cells) in all but 1 sample (Figure 1I), and the triple combination that included dexamethasone was synergistic in all samples (Figure 1J). No cytotoxicity was observed in normal peripheral blood mononuclear cells (supplemental Figure 1L).
On the basis of these data and previous data, combination therapy with venetoclax-dexamethasone can be more effective than venetoclax monotherapy (Scherr et al7 ; data not shown), so combination therapy was tested in vivo. We compared inotuzumab ozogamicin, venetoclax-dexamethasone, and the triple combination in Nod-Scid-IL2Rγnull (NSG) mice intravenously transplanted with 106 P020 (hyperdiploid), P021(t(1;19)), and L707 (t(17;19)) cells and monitored noninvasively by in vivo bioluminescence imaging (BLI) based on luciferase reporter expression.16 These genetic changes are rare in adult ALL, but t(17;19), which is found mainly in adolescents, has an extremely poor outcome.17 Upon engraftment confirmed with BLI, mice were treated with vehicle control or with venetoclax-dexamethasone, inotuzumab ozogamicin, or the triple combination, and leukemic burden, leukemia-free survival (LFS), and overall survival were monitored. On the basis of the different inotuzumab ozogamicin IC50’s, mice transplanted with the P020 and P021 models were treated with 10 µg/kg, whereas L707-engrafted mice were treated with 100 µg/kg, which is equivalent to human clinical dosing (supplemental Methods). In all 3 models, venetoclax-dexamethasone failed to induce CR with no survival benefit compared with controls (Figure 2A-D,G-H). In contrast, inotuzumab ozogamicin induced CR in 2 of 6 P021 mice but not in any P020 mice, despite a modest median survival benefit in both models (Figure 2A-D; data not shown). In contrast, triple combination therapy induced CR in all P020 and P021 mice and significantly prolonged median survival by 114 and 40 days, respectively. Dose dependency of inotuzumab ozogamicin therapy was next analyzed by treating P020 and P021 mice with 10-fold higher doses (100 µg/kg) (Figure 2E-F). All mice reached CR with inotuzumab ozogamicin monotherapy but eventually relapsed with a median survival of 140 days. In contrast, all mice survived BLI-negative upon treatment with the triple combination for up to 45 weeks after the start of therapy (Figure 2E-F). In the very-high-risk L707 model, the triple combination of inotuzumab ozogamicin and venetoclax-dexamethasone rapidly induced CR (no BLI signal was detectable) in all mice and prolonged median overall survival by 58 days, with 1 of 5 mice reaching long-term treatment-free LFS (Figure 2G-H). CD22 was expressed in all relapsed or refractory PDX samples analyzed, with expression levels depending on inotuzumab ozogamicin treatment in P021 cells, but no impairment of apoptosis initiated by inotuzumab ozogamicin and venetoclax was detectable (supplemental Figure 2A-G).
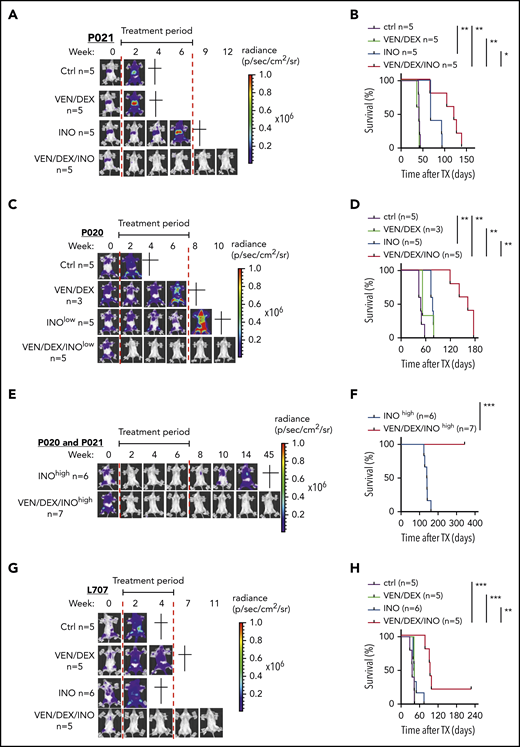
Effects of venetoclax, dexamethasone, and inotuzumab ozogamicin on ALL PDXs. (A-B) NSG recipients received 1 × 106 P021 cells intravenously. (A-B) Representative BLI results for weeks 0 to 12 after start of treatment (A) and Kaplan-Meier survival curve of recipients treated with venetoclax (20 mg/kg) and dexamethasone (1 mg/kg) 5 days per week and low-dose inotuzumab ozogamicin (10 µg/kg) once per week for 3 weeks as well as triple combination therapy (venetoclax-dexamethasone-inotuzumab ozogamicin) (B). Treatment (6 weeks) started upon engraftment confirmed by BLI 3 weeks after tumor inoculation as indicated (red lines). (C-D) NSG recipients received 1 × 106 P020 cells intravenously. Representative BLI results for weeks 0 to 10 after start of treatment (C) and Kaplan-Meier survival curves of recipients treated with venetoclax (20 mg/kg) and dexamethasone (1 mg/kg) 5 days per week for 6 weeks and low-dose inotuzumab ozogamicin (10 µg/kg) once per week for 3 weeks as well as triple combination therapy (venetoclax-dexamethasone-inotuzumab ozogamicin) (D). Treatment (6 weeks) started upon engraftment confirmed by BLI 3 weeks after tumor inoculation as indicated (red lines). (E-F) High-dose (100 µg/kg) inotuzumab ozogamicin (INOhigh) treatment of NSG mice inoculated with P021 and P020. Representative BLI results for weeks 0 to 45 after start of treatment with high-dose inotuzumab ozogamicin once per week for 3 weeks alone and in combination with venetoclax (20 mg/kg) and dexamethasone (1 mg/kg) 5 days per week for 6 weeks. (F) Kaplan-Meier survival curves of inotuzumab ozogamicinhigh (n = 6) (P021, n = 1; P020, n = 5) and venetoclax-dexamethasone-inotuzumab ozogamicinhigh (n = 7) (P021, n = 2; P020, n = 5). All mice treated with venetoclax-dexamethasone-inotuzumab ozogamicinhigh survived the whole investigation period of >50 weeks. (G-H) NSG recipients received 1 × 106 L707 cells intravenously. Tumor proliferation was monitored by noninvasive in vivo BLI based on luciferase reporter expression (G) and overall survival by Kaplan-Meier statistics (H). Representative BLI results for weeks 0 to 11 are shown in panel G. Treatment started upon engraftment confirmed by BLI 2 weeks after tumor inoculation. Recipients were treated with venetoclax (20 mg/kg) and dexamethasone (1 mg/kg) by oral gavage 5 days per week or with inotuzumab ozogamicin (100 µg/kg) intraperitoneally twice a week for 2 weeks or with triple combination therapy (venetoclax-dexamethasone-inotuzumab ozogamicin). Start and end of therapy are indicated by the red lines. Log-rank test was used for statistical survival analyses. *P < .05; **P < .01; ***P < .001.
Effects of venetoclax, dexamethasone, and inotuzumab ozogamicin on ALL PDXs. (A-B) NSG recipients received 1 × 106 P021 cells intravenously. (A-B) Representative BLI results for weeks 0 to 12 after start of treatment (A) and Kaplan-Meier survival curve of recipients treated with venetoclax (20 mg/kg) and dexamethasone (1 mg/kg) 5 days per week and low-dose inotuzumab ozogamicin (10 µg/kg) once per week for 3 weeks as well as triple combination therapy (venetoclax-dexamethasone-inotuzumab ozogamicin) (B). Treatment (6 weeks) started upon engraftment confirmed by BLI 3 weeks after tumor inoculation as indicated (red lines). (C-D) NSG recipients received 1 × 106 P020 cells intravenously. Representative BLI results for weeks 0 to 10 after start of treatment (C) and Kaplan-Meier survival curves of recipients treated with venetoclax (20 mg/kg) and dexamethasone (1 mg/kg) 5 days per week for 6 weeks and low-dose inotuzumab ozogamicin (10 µg/kg) once per week for 3 weeks as well as triple combination therapy (venetoclax-dexamethasone-inotuzumab ozogamicin) (D). Treatment (6 weeks) started upon engraftment confirmed by BLI 3 weeks after tumor inoculation as indicated (red lines). (E-F) High-dose (100 µg/kg) inotuzumab ozogamicin (INOhigh) treatment of NSG mice inoculated with P021 and P020. Representative BLI results for weeks 0 to 45 after start of treatment with high-dose inotuzumab ozogamicin once per week for 3 weeks alone and in combination with venetoclax (20 mg/kg) and dexamethasone (1 mg/kg) 5 days per week for 6 weeks. (F) Kaplan-Meier survival curves of inotuzumab ozogamicinhigh (n = 6) (P021, n = 1; P020, n = 5) and venetoclax-dexamethasone-inotuzumab ozogamicinhigh (n = 7) (P021, n = 2; P020, n = 5). All mice treated with venetoclax-dexamethasone-inotuzumab ozogamicinhigh survived the whole investigation period of >50 weeks. (G-H) NSG recipients received 1 × 106 L707 cells intravenously. Tumor proliferation was monitored by noninvasive in vivo BLI based on luciferase reporter expression (G) and overall survival by Kaplan-Meier statistics (H). Representative BLI results for weeks 0 to 11 are shown in panel G. Treatment started upon engraftment confirmed by BLI 2 weeks after tumor inoculation. Recipients were treated with venetoclax (20 mg/kg) and dexamethasone (1 mg/kg) by oral gavage 5 days per week or with inotuzumab ozogamicin (100 µg/kg) intraperitoneally twice a week for 2 weeks or with triple combination therapy (venetoclax-dexamethasone-inotuzumab ozogamicin). Start and end of therapy are indicated by the red lines. Log-rank test was used for statistical survival analyses. *P < .05; **P < .01; ***P < .001.
These data demonstrate the synergy of direct MOMP induction and DSB apoptosis signaling by concurrent treatment with venetoclax and inotuzumab ozogamicin in BCP-ALL. Indeed, only therapy with the combination of venetoclax-dexamethasone and inotuzumab ozogamicin induced CR in all 3 PDX models, with treatment-free long-term survival in all P020 and P021 mice being achieved in an inotuzumab ozogamicin dose-dependent manner. In contrast, dose-equivalent inotuzumab ozogamicin monotherapy failed to induce LFS corresponding to that in the INO-VATE ALL trial.2 Precise molecular mechanisms that mediate venetoclax and inotuzumab ozogamicin synergy and the contribution of dexamethasone to triple combination therapy remain to be determined. In addition, combined treatment may target different survival signals or threshold levels for the induction of apoptosis essential for ALL cells in vivo. Combination therapy with venetoclax, inotuzumab ozogamicin, and steroids should be evaluated in clinical trials of high-risk BCP-ALL patients either to optimize definitive pharmacotherapy or to deepen remission of leukemia before consolidation with cellular therapy.
For original data, please contact the corresponding author.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Andrea Hoffmann (Department of Orthopedic Surgery, Hannover Medical School) for providing mesenchymal stem cells and Jonathan Lühmann for conducting the optical genome mapping for retrospective cytogenetic analyses. The authors acknowledge Iris Dallmann for excellent technical help and the staff of the Central Animal Facility and Matthias Ballmaier from the Cell Sorting Core Facility (supported, in part, by the Braukmann-Wittenberg-Herz-Stiftung and the Deutsche Forschungsgemeinschaft) of Hannover Medical School.
This work was supported by H.W. and J. Hector-Stiftung und Stiftung Gerdes, and by a Cancer Research UK program grant (C27943/A12788) (O.H.).
Authorship
Contribution: H.K., M.E., and M.S. conceived the study and designed the experiments; H.K., K.B., U.K., C.S., S.E., S.R.T., and D.S. performed experiments and analyzed and interpreted the data; O.H., M.H., D.H.-K., and A.G. provided technical and material support; H.K., M.E., M.S., and O.H. wrote the manuscript; and all authors reviewed the manuscript before submission.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Matthias Eder, Department of Hematology, Hemostasis, Oncology and Stem Cell Transplantation, Hannover Medical School, Carl-Neuberg-Strasse 1, 30625 Hannover, Germany; e-mail: eder.matthias@mh-hannover.de; and Michaela Scherr, Department of Hematology, Hemostasis, Oncology and Stem Cell Transplantation, Hannover Medical School, Carl-Neuberg-Strasse 1, 30625 Hannover, Germany; e-mail: scherr.michaela@mh-hannover.de.