

Key Points
B-PLL is tightly linked to MYC aberrations (translocation or gain) and 17p (TP53) deletion.
Cases of B-PLL with MYC aberration and 17p (TP53) deletion have the worst prognosis.

Abstract
B-cell prolymphocytic leukemia (B-PLL) is a rare hematological disorder whose underlying oncogenic mechanisms are poorly understood. Our cytogenetic and molecular assessments of 34 patients with B-PLL revealed several disease-specific features and potential therapeutic targets. The karyotype was complex (≥3 abnormalities) in 73% of the patients and highly complex (≥5 abnormalities) in 45%. The most frequent chromosomal aberrations were translocations involving MYC [t(MYC)] (62%), deletion (del)17p (38%), trisomy (tri)18 (30%), del13q (29%), tri3 (24%), tri12 (24%), and del8p (23%). Twenty-six (76%) of the 34 patients exhibited an MYC aberration, resulting from mutually exclusive translocations or gains. Whole-exome sequencing revealed frequent mutations in TP53, MYD88, BCOR, MYC, SF3B1, SETD2, CHD2, CXCR4, and BCLAF1. The majority of B-PLL used the IGHV3 or IGHV4 subgroups (89%) and displayed significantly mutated IGHV genes (79%). We identified 3 distinct cytogenetic risk groups: low risk (no MYC aberration), intermediate risk (MYC aberration but no del17p), and high risk (MYC aberration and del17p) (P = .0006). In vitro drug response profiling revealed that the combination of a B-cell receptor or BCL2 inhibitor with OTX015 (a bromodomain and extra-terminal motif inhibitor targeting MYC) was associated with significantly lower viability of B-PLL cells harboring a t(MYC). We concluded that cytogenetic analysis is a useful diagnostic and prognostic tool in B-PLL. Targeting MYC may be a useful treatment option in this disease.
Medscape Continuing Medical Education online
In support of improving patient care, this activity has been planned and implemented by Medscape, LLC and the American Society of Hematology. Medscape, LLC is jointly accredited by the Accreditation Council for Continuing Medical Education (ACCME), the Accreditation Council for Pharmacy Education (ACPE), and the American Nurses Credentialing Center (ANCC), to provide continuing education for the healthcare team.
Medscape, LLC designates this Journal-based CME activity for a maximum of 1.00 AMA PRA Category 1 Credit(s)™. Physicians should claim only the credit commensurate with the extent of their participation in the activity.
Successful completion of this CME activity, which includes participation in the evaluation component, enables the participant to earn up to 1.0 MOC points in the American Board of Internal Medicine's (ABIM) Maintenance of Certification (MOC) program. Participants will earn MOC points equivalent to the amount of CME credits claimed for the activity. It is the CME activity provider's responsibility to submit participant completion information to ACCME for the purpose of granting ABIM MOC credit.
All other clinicians completing this activity will be issued a certificate of participation. To participate in this journal CME activity: (1) review the learning objectives and author disclosures; (2) study the education content; (3) take the post-test with a 75% minimum passing score and complete the evaluation at http://www.medscape.org/journal/blood; and (4) view/print certificate. For CME questions, see page 1879.
Disclosures
Associate Editor John C. Byrd received grants for clinical research from Acerta; Genentech, Inc.; Janssen Pharmaceuticals, Inc.; and Pharmacyclics, Inc. CME questions author Laurie Barclay, freelance writer and reviewer, Medscape, LLC and the authors declare no competing financial interests.
Learning objectives
Upon completion of this activity, participants will be able to:
Describe the genetic portrait of B-cell prolymphocytic leukemia (B-PLL), according to cytogenetic and molecular testing in a case series
Determine correlations between cytogenetic and molecular findings and patients' clinical outcomes, according to cytogenetic and molecular testing in a case series
Identify primary B-PLL cells' in vitro response to novel targeted therapeutics, according to cytogenetic and molecular testing in a case series
Release date: November 21, 2019; Expiration date: November 21, 2020
Introduction
B-cell prolymphocytic leukemia (B-PLL) is a very rare disease that accounts for <1% of all cases of chronic B-cell leukemia and generally occurs in elderly individuals. According to the World Health Organization criteria, B-PLL can be diagnosed when prolymphocytes comprise >55% of the lymphoid cells in peripheral blood. Diagnosis on the basis of clinical and morphological data can be difficult because B-PLL shares a number of features with other B-cell malignancies, including splenic marginal zone lymphoma (MZL), mantle cell lymphoma (MCL), and chronic lymphocytic leukemia (CLL). For example, an unusual leukemic form of MCL mimicking B-PLL is distinguished by the presence of the t(11;14)(q13;q32) translocation.1,2 B-PLL does not have a specific immunophenotype and has low Matutes scores,3,4 as observed for other B lymphoproliferative disorders.
Little is known about the oncogenic events leading to the development and clinical heterogeneity of B-PLL. Given the disease’s rarity, its cytogenetic features have only been described in case reports and small series,5,6 and large-scale genomic sequencing has not yet been performed for this disease.
Rituximab-based chemo-immunotherapy is often used to treat patients who have B-PLL but no TP53 aberrations. In patients with TP53 aberrations, alemtuzumab-based regimens have been implemented. Lasting treatment responses are rare, and the prognosis is generally poor.7 Recent data from case reports and small series suggest that 2 agents targeting B-cell signaling (ibrutinib and idelalisib) are efficacious.8-10 Hence, allogeneic hematopoietic stem cell transplantation is still the only curative treatment of B-PLL, albeit in younger patients only.7
In the present study, we performed cytogenetic and molecular assessments in one of the largest reported series of patients with a validated diagnosis of B-PLL. We further sought correlations between the cytogenetic and molecular findings and the patients’ clinical outcomes. Lastly, we studied the in vitro response of the primary B-PLL cells to novel targeted therapeutic agents. To the best of our knowledge, the present study is the first to provide an extensive genetic portrait of B-PLL and to offer insights into prognostic markers and treatment options.
Patients, materials, and methods
Patient selection
Databases from 28 French health care establishments were retrospectively screened for cases of de novo B-PLL. Patients with a history of another B-cell malignancy (CLL or MZL) were excluded. A total of 87 patients with an available blood smear at diagnosis were first identified locally, based on cell morphological criteria. All 87 blood smears were blind-reviewed by 3 independent expert cytologists (L.B., K.M., and C.S.), and 10 representative smears were subsequently reviewed further by a fourth cytologist (J.-F.L.) (supplemental Methods; supplemental Table 1; available on the Blood Web site). Only 34 of the 87 cases met the World Health Organization criteria for B-PLL (ie, prolymphocytes accounted for >55% of the lymphoid cells in peripheral blood). A diagnosis of MCL was ruled out according to karyotype (K) and fluorescence in situ hybridization (FISH) assays: no CCND1 rearrangements or other infrequent translocations involving CCND2 and CCND3 were observed.11,12 Four other patients carrying a translocation t(11;14)(q13;q32) with a CCND1 rearrangement were diagnosed as having MCL and were excluded. Cytogenetic and molecular analyses were performed on the available material: at diagnosis for 21 patients, during follow-up and before treatment for 10 patients (median time between diagnosis and sampling, 44 months), and at relapse for 3 patients. The study was performed in accordance with the Declaration of Helsinki and was approved by the local investigational review board (CPP-Ile-de-France VI, Paris, France) on 21 May 2014.
Karyotyping and FISH analysis
Standard chromosome banding analyses were used to obtain R- or G-banded chromosomes from peripheral blood (n = 28), bone marrow (n = 4), or spleen (n = 1) samples. The samples were cultured for 48 to 72 hours with 12-O-tetradecanoylphorbol-13-acetate (n = 6) or CpG-oligonucleotides and interleukin-2 (n = 27). All the karyotyping results were reviewed by the members of the Groupe Francophone de Cytogénétique Hématologique and classified according to the International System for Human Cytogenetic Nomenclature (2016). Complex Ks (CKs) and highly complex Ks (HCKs) were defined as the presence of at least 3 or 5 numerical or structural chromosomal abnormalities, respectively. Standard FISH was performed in all cases. The specific probes and procedures used are detailed in the supplemental Methods.
Whole-exome sequencing
DNA extracted from flow-sorted CD19+ tumor cells (and CD5+, IGL, or IGK when appropriate) and nontumor (CD3+) cells (considered to be germinal controls) was used for exome capture according to standard procedures. Paired-end sequencing was performed by using HiSeq2000 sequencing instruments (Illumina, San Diego, CA). More detailed information can be found in the supplemental Methods.
Targeted deep resequencing
Mutation validation using targeted deep resequencing was performed as previously described.13 Details are available in the supplemental Methods.
IGHV analysis
IGH rearrangement sequencing was performed on DNA extracted from sorted tumor cells or from whole blood, as previously described.14 Depending on the percentage of identity of the IGHV genes with their germline counterparts, sequences were considered as unmutated (100% identity), minimally/borderline mutated (99.9%-97%), or significantly mutated (<97% identity), following criteria proposed by Hadzidimitriou et al15 for MCL and Bikos et al16 for MZL.
RNA sequencing
RNA sequencing (RNA-seq) was performed on high-quality RNA from twelve B-PLL as previously described.17 More detailed information can be found in the supplemental Methods.
In vitro cell viability and programmed cell death assays
The viability of primary B-PLL cells was assessed with the adenosine triphosphate (ATP)-based CellTiter-Glo 2.0 assay (Promega, Madison, WI). Briefly, after pretreatment (or not) with the bromodomain and extra-terminal motif protein inhibitors (iBETs) OTX015 or JQ1 (500 nM), B-PLL cells were exposed for 48 hours to increasing concentrations of fludarabine, ibrutinib, idelalisib, or venetoclax. Viability was determined by normalizing luminescence units against a dimethyl sulfoxide control. Alternatively, programmed cell death was measured by flow cytometry using Annexin-V and propidium iodide colabeling, after B-PLL cell treatment of 48 hours with ibrutinib, idelalisib, venetoclax, or OTX015. Details are provided in the supplemental Methods. All drugs were purchased from Selleckchem (Houston, TX).
Statistical analysis
Overall survival (OS) was defined as the time interval between diagnosis and death or last follow-up. Categorical variables were compared by using a χ2 test or Fisher’s exact test; continuous variables were compared by using the Mann-Whitney U test. Survival analyses were performed by using the Kaplan-Meier method, and the log-rank test was used for intergroup comparisons of OS curves. Two-sided P < .05 was considered statistically significant. All statistical analyses were performed by using MedCalc software (version 17.8.6, Ostend, Belgium).
Additional methods are described in the supplemental Methods.
Results
Characteristics of the study population
The study included 34 patients diagnosed with de novo B-PLL between 1992 and 2017 (Table 1). There was a predominance of male subjects (59%), and the median (range) age at diagnosis was 72 (46.2-87.9) years. The median lymphocyte count was 36 (4.6-244) G/L, and the median proportion of prolymphocytes was 79.5 (60-100). Nineteen of the 34 patients (56%) were positive for CD5 (supplemental Table 2). The median follow-up time after diagnosis was 47 (0.2-141) months. Most of the patients had been treated (29 of 33 [88%]) with a median time after diagnosis of 3.2 (0-106) months.
Chromosomal aberrations: MYC is the most frequently altered gene in B-PLL
Karyotyping was performed in 33 patients (Table 1; supplemental Table 3). A CK was found in 73% of the patients, and HCK in 45%. By combining K and FISH data, we observed that the most frequent abnormality, found in 21 (62%) of the 34 patients, was a translocation involving the MYC gene [t(MYC)]. This translocation involved the IGH locus in 12 cases, IGL in 6, and IGK in 1; the MYC gene partner was not identified in 2 cases. Five (15%) of the 34 patients had gained copies of the MYC gene: three had 3 copies, one had 4 copies, and one had MYC amplification. Thus, the majority of the cases (26 of 34 [76%]) had an MYC abnormality, MYC translocation and MYC gain being mutually exclusive. The other frequent chromosomal aberrations were: deletion (del)17p including the TP53 gene (13 of 34 [38%]), trisomy (tri) 18/18q (10 of 33 [30%]), del13q (10 of 34 [29%]), tri3 (8 of 33 [24%]), tri12 (8 of 34 [24%]), and del8p (7 of 31 [23%]) (Figure 1).
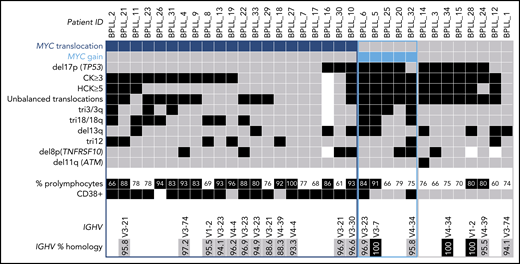
Distribution of chromosomal abnormalities detected in 34 patients with B-PLL with 3 cytogenetic subgroups of patients: MYC translocation, MYC gain, and no MYC aberration. Each column represents 1 patient, and each row 1 particular genetic or laboratory parameter. Color code: black or blue, presence; gray, absence; white, not available. CK, ≥3 chromosomal abnormalities; HCK, ≥5 chromosomal abnormalities. The percentages of prolymphocytes (indicated in black boxes) correspond to above-median values, relative to the cohort as a whole.
Distribution of chromosomal abnormalities detected in 34 patients with B-PLL with 3 cytogenetic subgroups of patients: MYC translocation, MYC gain, and no MYC aberration. Each column represents 1 patient, and each row 1 particular genetic or laboratory parameter. Color code: black or blue, presence; gray, absence; white, not available. CK, ≥3 chromosomal abnormalities; HCK, ≥5 chromosomal abnormalities. The percentages of prolymphocytes (indicated in black boxes) correspond to above-median values, relative to the cohort as a whole.
An analysis of copy number values (calculated from the whole-exome sequencing [WES] coverage in 16 patients) confirmed the majority of recurrent losses and gains observed by karyotyping and/or FISH analyses and highlighted other novel recurrent copy number variations: losses of 5q22-23, 9q34, 14q24, and 19p13, and gains of 17q24, 1q31-42, and 4q27-35 (supplemental Table 4).
IGHV analyses: preferential usage of VH3 and VH4 genes, and frequently mutated IGHV genes
We obtained clonal IGH sequences in 19 patients with B-PLL. In 17 (89%) of the 19 B-PLL cases, a member of the IGHV3 (n = 11, including 4 IGHV3-23) or IGHV4 (n = 6) subgroups was involved; the IGHV1 subgroup gene (IGHV1-2) was used in the 2 remaining cases. Furthermore, the vast majority of B-PLL sequences (15 of 19 [79%]) displayed significantly mutated IGHV genes (Figure 1; supplemental Table 5).
The spectrum of somatic mutations
WES of paired tumor-control DNA was performed in 16 patients, and mutations were validated by using targeted deep-resequencing and/or RNA-seq (supplemental Figure 1; supplemental Table 6). We identified 10 genes that were mutated in at least 2 patients (Table 2). The most frequently mutated gene was TP53, with a total of 7 mutations observed in 6 (38%) of the 16 patients. These TP53 mutations were predicted to affect the protein’s DNA-binding domain (supplemental Figure 2). In all cases, these mutations were associated with a del17p. Mutations in MYD88 were found in 4 patients. Interestingly, the MYD88 mutation was associated with tri3 in 2 cases, with a variant allele frequency of 66.5% (BPLL_6) and 67.4% (BPLL_32) indicating that the mutated copy was duplicated. Mutations in genes that have a role in chromatin modifications were recurrent, including the X-linked BCOR gene encoding a BCL6 corepressor (n = 4), CHD2 (n = 2), SETD2 (n = 2), and other genes mutated in 1 patient each (CREBBP, EZH2, KMT2D, NCOR1, SETD1B, and TET2). The MYC gene had missense mutations in 3 patients, all of whom harbored a t(MYC). Two mutations were just downstream of the transactivation domain, and one was in the bHLH domain. The products of other relevant mutated genes were found to be involved in RNA metabolism (SF3B1, BCLAF1, ZRSR2, and WBP4), cell migration (CXCR4), cell cycle (CDKN3 and RB1), apoptosis (TP73), and NOTCH signaling (NOTCH2 and FBXW7) (Figure 2).
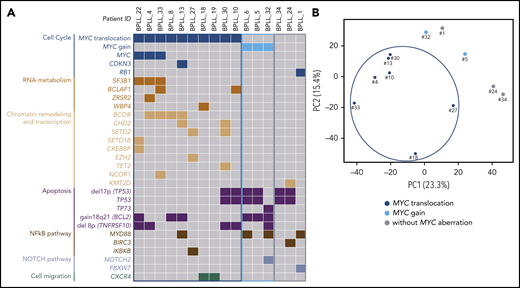
WES and RNA-seq. (A) Distribution of the chromosomal aberrations and mutations in the 3 cytogenetic subgroups (MYC translocation, MYC gain, and no MYC aberration) in the 16 cases analyzed by using WES. Chromosomal abnormalities: gray, absence; color, presence. Mutations: gray, wild type; color, mutated. (B) Principal component analysis of differential gene expression patterns for 12 samples with various cytogenetic abnormalities.
WES and RNA-seq. (A) Distribution of the chromosomal aberrations and mutations in the 3 cytogenetic subgroups (MYC translocation, MYC gain, and no MYC aberration) in the 16 cases analyzed by using WES. Chromosomal abnormalities: gray, absence; color, presence. Mutations: gray, wild type; color, mutated. (B) Principal component analysis of differential gene expression patterns for 12 samples with various cytogenetic abnormalities.
IGL/PVT1 fusions
Using RNA-seq, we detected IG fusion transcripts in 3 of 7 patients with t(MYC). In case #27 harboring a t(8;14)(q24;q32), IGHG1 was fused with intron 1 of MYC. In cases BPLL_4 and BPLL_30 with a t(8;22)(q24;q11), the PVT1 gene was fused to IGLL5 or IGLV4-69*01, respectively. The 2 latter fusions were confirmed by using a reverse-transcriptase polymerase chain reaction assay. No other recurrent fusion transcripts were found in the 12 patients with RNA-seq data (supplemental Table 7).
Clonal organization
With a view to identifying early and late oncogenic events, we evaluated the clonal or subclonal nature of chromosomal abnormalities and recurrent somatic mutations. We observed that t(MYC), del17p, tri12, and mutations in the TP53, BCOR, MYC, SF3B1, and MYD88 genes were mostly clonal and should be considered as early events in B-PLL. In contrast, MYC gain, del8p, and del13q were frequently subclonal, indicating that they are acquired (supplemental Figure 3).
Correlations between cytogenetic aberrations, mutations, and gene expression patterns
Three cytogenetic subgroups were identified (Figure 1; Table 3). The main subgroup of patients (21 of 34 [62%]) had a t(MYC). Relative to the patients without t(MYC), this subgroup had a higher percentage of prolymphocytes (P = .03), a higher proportion of CD38+ cells (P < .0001), a lower level of cytogenetic complexity (P = .0005), a lower proportion of cases with HCK (P = .0004), fewer unbalanced translocations (P = .04), and fewer cases with del17p (P = .0006). Mutations in MYC and in genes involved in RNA metabolism, chromatin remodeling and transcription were almost exclusively observed in the t(MYC) subgroup, whereas mutations in TP53 were less frequent (Figure 2A). A principal component analysis of gene expression data from the 12 cases analyzed by using RNA-seq showed that the 7 patients with t(MYC) clustered together (Figure 2B). Taken as a whole, these results suggest that patients with t(MYC) form a homogeneous subgroup of B-PLL. A second subgroup of B-PLL with MYC gain (5 of 34 [15%]) was associated with HCK (P = .01) and tri3 (P = .008). The remaining 8 patients corresponded to a third subgroup lacking an MYC aberration. Compared with patients with the MYC aberration, this third subgroup had a lower percentage of prolymphocytes (P = .03), no CD38+ expression (P < .001), a higher number of chromosomal aberrations (P = .02), and a higher frequency of del17p (P = .03) (Table 3).
Survival analyses: the prognostic values of MYC aberrations and del17p
The median OS for the entire study cohort was 125.7 months. Patients with t(MYC) had a median (95% CI) OS of 57.5 months (25.7-132.1), which did not differ significantly from the value observed for patients with MYC gain (median, 66.5 months) (Figure 3A). Hence, we pooled patients with an MYC aberration for subsequent OS comparisons and found that they had a significantly shorter OS than patients without MYC aberrations (P = .03) (Figure 3B).
![Overall survival in patients with B-PLL. Panels A-B as a function of MYC aberrations [t(MYC) or gain] and the presence or absence of MYC aberration; the t(MYC) and MYC gain data were pooled. (A) t(MYC) median (95% CI), 57.5 months (25.7-132.1); MYC gain median (95% CI), 66.5 months (4.7-undetermined). (B) MYC aberration [t(MYC) + MYC gain] median (95% CI), 57.5 (25.7-132.1). (C) MYC and del17p status. MYC aberration without del17p median (95% CI), 125.7 months (52.2-132.1); MYC aberration with del17p median (95% CI), 11.1 months (4.7-66.5). ab, aberration.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/134/21/10.1182_blood.2019001187/7/m_bloodbld2019001187f3.png?Expires=1765187030&Signature=DXgNKxZiwSDBHvHUFpI-FdaACzHVSRMANLNkrb3dTwLuejTz5eqw4oFK51XqwoY2XETndsY9HblAJkUk-NGG~TTGpathm2Rap3vat-uDLcTx8DbYJVsjS8waKMGrdFqzK8RQDYierzbFpFA6GSt5tYmEMjjaSlpoFURVZYoTXOfXp1QHM8~IXyhBldRirn9ivgY~BHReG6hsTtxSD2LROyaHqLFVvrmE1BtWkuWk1htX1CecwIP3xEVVMN85wZgDiDZl33WMv-0NBtCIoyZPlwkVzh5iTi914IVqn8ixGToOKemlsw2JEILsjmc8xzy~Pwo8GbnudVpHh3m2IiYi4g__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Overall survival in patients with B-PLL. Panels A-B as a function of MYC aberrations [t(MYC) or gain] and the presence or absence of MYC aberration; the t(MYC) and MYC gain data were pooled. (A) t(MYC) median (95% CI), 57.5 months (25.7-132.1); MYC gain median (95% CI), 66.5 months (4.7-undetermined). (B) MYC aberration [t(MYC) + MYC gain] median (95% CI), 57.5 (25.7-132.1). (C) MYC and del17p status. MYC aberration without del17p median (95% CI), 125.7 months (52.2-132.1); MYC aberration with del17p median (95% CI), 11.1 months (4.7-66.5). ab, aberration.
Overall survival in patients with B-PLL. Panels A-B as a function of MYC aberrations [t(MYC) or gain] and the presence or absence of MYC aberration; the t(MYC) and MYC gain data were pooled. (A) t(MYC) median (95% CI), 57.5 months (25.7-132.1); MYC gain median (95% CI), 66.5 months (4.7-undetermined). (B) MYC aberration [t(MYC) + MYC gain] median (95% CI), 57.5 (25.7-132.1). (C) MYC and del17p status. MYC aberration without del17p median (95% CI), 125.7 months (52.2-132.1); MYC aberration with del17p median (95% CI), 11.1 months (4.7-66.5). ab, aberration.
With regard to the combination of MYC and del17p aberrations, 3 distinct prognostic groups were identified with significant differences in OS (P = .0006) (Figure 3C). The patients without an MYC aberration (n = 8) had the lower risk (median not reached). Patients with an MYC aberration (translocation or gain) but no del17p (n = 18) had an intermediate risk (125.7 months). The high-risk group corresponded to patients with both MYC and del17p (n = 7; 11.1 months).
Other chromosomal abnormalities and K complexity had no significant impact on OS. Patients with del17p tended to have a shorter OS than those without del17p (supplemental Figure S4).
In vitro drug response assays
To evaluate the sensitivity of B-PLL cells to drugs used in the treatment of B-cell malignancies, we first used a cell viability assay based on the quantification of cellular ATP levels (an indicator of metabolic activity18 ) on primary B-PLL cells after 48 hours of exposure to ibrutinib, idelalisib (both B-cell receptor inhibitors), venetoclax (a highly specific inhibitor of the antiapoptotic protein BCL2), and fludarabine. The drugs were tested alone or in combination with OTX015 (an iBET known to suppress MYC expression in leukemia and lymphoma cell lines).19-21 Treatment with increasing doses of single drugs resulted in reduced cell viability in the 3 cases with t(MYC) (BPLL_8, BPLL_13, and BPLL_18) but varied from one drug to another and from one patient to another. Relative to treatments with single drugs, OTX015 cotreatment with low doses of fludarabine, ibrutinib, or venetoclax was associated with a much lower level of viability (<30%) in patients with t(MYC) (Figure 4A). In contrast, the patient harboring a del17p but not an MYC aberration (BPLL_34) was significantly more resistant to single drugs and OTX015 cotreatments than patients with t(MYC) (Figure 4B).
![The combination of drugs frequently used to treat B-cell malignancies with iBETs enhances the killing of primary B-PLL cells with t(MYC). (A) B-PLL cells from 3 patients with t(MYC) were treated with the indicated concentrations of ibrutinib, idelalisib, venetoclax, or fludarabine in the presence or absence of OTX015 (500 nM). Viability was assessed with an ATP-based CellTiter-Glo 2.0 kit. (B) B-PLL cell viability was measured and analyzed as in panel A in a patient with del17p but no MYC aberration. (C) Cell death was quantified in primary B-PLL cells from patient BPLL_8 [with t(MYC)] with or without pretreatment with OTX015 (500 nM) and exposure for 48 hours to ibrutinib (7.5 µM), idelalisib (50 µM), or venetoclax (10 nM). The percentages refer to Annexin-V–positive or Annexin-V–/propidium iodine–positive cells. (D) Primary B-PLL cells from BPLL_34 (with del17p but no MYC aberration) were treated with drugs and analyzed as in panel C. Bars represent the mean ± SEM.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/134/21/10.1182_blood.2019001187/7/m_bloodbld2019001187f4.png?Expires=1765187030&Signature=5DqDKBO2japrh1HPUI8aP7Gtxeyf8gqmYizSnLBrnUlg-3-6hZXW0JA~pX7~y9wuD1kgK17R436nfD5~ISLcnYRTHm9eFzL5HJFgEFBfU9RnOoDMjCP-9PpfsRrBhJuRHXjf4HKQL5aNFTmfoeb2yxytNv-ZaxpqGZTVRG1LLsf2H0TPC8clfnLx12WbohfrGPT34Y~CJjxS-OpS2twMRk1XCCimTJPQeDCeiZC3RQ4B0Q9jEnUixvDiN4GElH-i8E253sT-NE3rRE1tXZcWvXjesVd4KMv-RL0AgLKKGOwmh1F5Lu3p-x-I~UtmN2eAXuwdcb-sHFfgQjwrLvnGYw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
The combination of drugs frequently used to treat B-cell malignancies with iBETs enhances the killing of primary B-PLL cells with t(MYC). (A) B-PLL cells from 3 patients with t(MYC) were treated with the indicated concentrations of ibrutinib, idelalisib, venetoclax, or fludarabine in the presence or absence of OTX015 (500 nM). Viability was assessed with an ATP-based CellTiter-Glo 2.0 kit. (B) B-PLL cell viability was measured and analyzed as in panel A in a patient with del17p but no MYC aberration. (C) Cell death was quantified in primary B-PLL cells from patient BPLL_8 [with t(MYC)] with or without pretreatment with OTX015 (500 nM) and exposure for 48 hours to ibrutinib (7.5 µM), idelalisib (50 µM), or venetoclax (10 nM). The percentages refer to Annexin-V–positive or Annexin-V–/propidium iodine–positive cells. (D) Primary B-PLL cells from BPLL_34 (with del17p but no MYC aberration) were treated with drugs and analyzed as in panel C. Bars represent the mean ± SEM.
The combination of drugs frequently used to treat B-cell malignancies with iBETs enhances the killing of primary B-PLL cells with t(MYC). (A) B-PLL cells from 3 patients with t(MYC) were treated with the indicated concentrations of ibrutinib, idelalisib, venetoclax, or fludarabine in the presence or absence of OTX015 (500 nM). Viability was assessed with an ATP-based CellTiter-Glo 2.0 kit. (B) B-PLL cell viability was measured and analyzed as in panel A in a patient with del17p but no MYC aberration. (C) Cell death was quantified in primary B-PLL cells from patient BPLL_8 [with t(MYC)] with or without pretreatment with OTX015 (500 nM) and exposure for 48 hours to ibrutinib (7.5 µM), idelalisib (50 µM), or venetoclax (10 nM). The percentages refer to Annexin-V–positive or Annexin-V–/propidium iodine–positive cells. (D) Primary B-PLL cells from BPLL_34 (with del17p but no MYC aberration) were treated with drugs and analyzed as in panel C. Bars represent the mean ± SEM.
To confirm these results, a complementary cytofluorometric approach was used. In BPLL_8 with t(MYC), treatments with drugs alone killed between 13% and 27% of the cells. An additive effect of OTX015 was observed; between 48% and 63% of the cells were killed (Figure 4C). Cells from patient BPLL_34 (del17p but no MYC aberration) were less sensitive to all drugs tested in both the presence and absence of OTX015 (Figure 4D). Thus, the ATP assay and flow cytometry assay results highlighted the efficacy of combined drug + iBET treatment of cells from patients with t(MYC). The enhancing effect of the iBETs on the programmed cell death induced by fludarabine, ibrutinib, and venetoclax was fully corroborated by the results of cotreatment with JQ1, another well-known iBET (supplemental Figure 5B-C). It should be noted that we did not test any cases with MYC gain.
Discussion
In the present study, 34 patients with B-PLL were genetically characterized. Given that the diagnosis of B-PLL is challenging, a thorough morphological review of blood smears was performed by 3 independent cytologists. Although patients with a history of MZL were excluded, the absence of a biopsy analysis means that we cannot fully rule out the possible presence of MZL with a prolymphocytoid transformation, an extremely rare situation.22
To the best of our knowledge, the present series is the largest yet to have undergone a comprehensive genomic characterization of B-PLL. We found that the most frequent aberrations are related to MYC alterations (in 76% of cases), highlighting the major role of MYC in the pathogenesis of B-PLL. Strikingly, MYC translocations and MYC gain were found to be mutually exclusive. Translocations in which the MYC gene is repositioned under the control of an enhancer (usually immunoglobulin gene enhancers) result in the overexpression of MYC, as does the gain of one or more copies of MYC.23 Although MYC translocations and mutations are classically associated with Burkitt lymphoma (BL), aberrations in MYC have also been linked to almost all B-cell neoplasms. With regard to B-PLL, MYC translocations and MYC gain have been reported in a few cases.24-29 In the present series, t(MYC) was present in the major clone, but MYC gain was mainly subclonal and/or overlooked as part of a CK. Thus, performing FISH with a specific probe is essential to identify MYC gain or amplification. MYC gene mutations were found in 3 cases, all of which also had t(MYC). These mutations were not located in the transactivation domain (the hot spot for MYC mutations in BL). However, it is likely that MYC mutations have a role in the physiopathology of B-PLL (as they do in BL) by enhancing the oncogenicity of Myc.30 We found a fusion transcript between PVT1 and an IGL gene in 2 patients with a variant MYC translocation t(8;22). This fusion subsequently disrupted the PVT1 gene, a long intergenic noncoding RNA located 55 kb downstream of MYC and which has been identified as a recurrent breakpoint in BL harboring variant translocations t(2;8) or t(8;22).31,32 In a recent study, the PVT1 promoter was found to behave like a tumor-suppressor DNA element: silencing the PVT1 promoter increased MYC transcription and cell proliferation.33 As with MYC mutations, disruption of the PVT1 gene in some patients with B-PLL might cooperate functionally with translocations to increase MYC-mediated oncogenesis.
Our present results confirmed previous case reports in which a CK was frequent in B-PLL.24,27,34 The frequency of a CK in B-PLL (73% in the present study) is higher than that reported in BL (34%),35 MCL (40%-60%),36,37 MZL (53%),38 and CLL at diagnosis (15%)39-42 and is similar to that observed in high-risk CLL.43 Whereas t(MYC) is the main abnormality in B-PLL (62%), it is rare in other chronic B-cell disorders (ie, <5% in MCL, MZL, and CLL).28,38,44 The frequency of del17p in B-PLL (38%) observed here is higher than for MCL and MZL (both ∼20%)37,38 or for CLL (5%-7% at diagnosis).39,45 In addition to MYC and TP53 abnormalities, several other recurrent chromosomal aberrations were found in B-PLL, including trisomies 3, 12, and 18, del13q, and del8p. None of the chromosomal abnormalities observed in B-PLL are specific and thus can be observed in other mature B-cell malignancies, albeit at different frequencies. Similarly, all the driver genes recurrently mutated in B-PLL are also involved in other chronic B-cell disorders but not with the same prevalence. In line with previous studies,5,6 we found a high frequency of TP53 mutations (38%) in B-PLL; this frequency is higher than that observed for other B-cell malignancies such as MCL (18%),46 LZM (14%),47 CLL (10%),48 and BL (24%).49
We identified a high frequency of clonal BCOR mutations, which were present only in t(MYC) patients. BCOR encodes a Bcl6 co-repressor that silences BCL6 targets by epigenetic modifications.50 Inactivating mutations of BCOR have been found in myeloid disorders51,52 but are very unusual in B-cell diseases, except in the rare splenic diffuse red pulp lymphoma.53 The association of BCOR mutations and t(MYC) might be a specific feature of B-PLL. Indeed, a recent study of Eμ-Myc lymphomas in a mouse model revealed a high frequency of Bcor mutations and showed that Bcor was a cooperative tumor suppressor gene, the disruption of which can accelerate Myc-driven lymphomagenesis.54
Our data showed that patients with B-PLL and t(MYC) form a homogeneous subgroup, with a less K complexity, a higher proportion of prolymphocytes, and a higher frequency of CD38 expression. Interestingly, clonal mutations in the MYC, BCOR, and SF3B1 genes clustered together in some patients with t(MYC), suggesting oncogenic cooperation in the development of B-PLL. MYC aberration is a feature shared with BL; however, in contrast to BL, no mutation was found in TCF3, ID3, or CCND3 genes in B-PLL.49
van der Velden et al55 have suggested that B-PLL is a specific subgroup of t(11;14)-negative MCL, based on similarities in the immunophenotype, immunogenotype, and gene expression profile. We did not find any CCND1 or ATM mutations (the most frequent mutations in MCL56 ) in our patients with B-PLL. The vast majority of B-PLL expressed significantly mutated IGHV genes belonging to the IGHV3 and IGHV4 subgroups, in keeping with previous reports.57,58 This finding contrasts with that of MCL, which exhibits predominantly no or minimal somatic hypermutations.15
Taken as a whole, our data show that the genomic profile of B-PLL shares similarities with that of chronic B-cell malignancies but also displays unique combinations of alterations, thus confirming that B-PLL is a distinct entity.
The median OS time in the study cohort was 125.7 months, longer than the times reported in 2 earlier studies.6,59 This difference might be due to our patients’ treatments with rituximab, alemtuzumab, or fludarabine, all of which are reportedly efficacious in B-PLL.60,61 In light of our results, we propose a hierarchical prognostic model that depends on MYC aberration (translocation or gain) and del17p. Patients without an MYC aberration have indolent disease (regardless of their 17p status), whereas patients with an MYC aberration have a significantly shorter OS time. A combination of MYC aberration and del17p confers the worse outcome, with a median OS of <1 year. By analogy with the “double-hit” form of CLL involving del17p and MYC gain,43 these patients may be considered as having a very-high-risk form of B-PLL. Due to the rarity of this disease, our data were obtained with a relatively small number of patients and thus need to be confirmed in future studies.
Our in vitro data showed that a combination of ibrutinib, idelalisib, or venetoclax with OTX015 is more cytotoxic than the drugs used alone, especially in patients with MYC translocation. This scenario is concordant with the known effects of OTX015, an inhibitor of the bromodomain-containing proteins BRD2, BRD3, and BRD4. These proteins act as epigenetic readers and have an important role in the regulation of transcription and the modulation of oncogene expression. As with other iBETs, OTX015 strongly downregulates the transcription of genes regulated by super-enhancers such as MYC.20 Current approaches to the treatment of B-PLL (as with CLL) are based on del17p. However, our results strongly suggest that MYC should be taken into account and could be targeted. OTX015 is already in phase 1 clinical studies with a relatively good safety profile62 ; when combined with targeted drugs, it might constitute an epigenetic treatment option in patients with B-PLL and MYC translocation. In any case, further investigations with a larger number of patients (especially those with MYC gain) are required to make progress in this field.
In conclusion, we found that patients with B-PLL have CKs and HCKs, a high frequency of MYC aberration (by translocation or gain), frequent del17p, and frequent mutations in the TP53, BCOR, MYD88, and MYC genes. We identified 3 prognostic subgroups, depending on MYC and 17p status. Patients with both MYC aberration and del17p had the shortest OS and should be considered as a high-risk subgroup. These results showed that cytogenetic analysis is a useful diagnostic and prognostic tool in B-PLL. We recommend karyotyping and FISH (for MYC and TP53) analyses whenever B-PLL is suspected. Moreover, these in vitro data strongly suggest that drugs targeting the BCR and BCL2 in combination with an iBET might be a treatment option for patients with B-PLL.
The WES and RNA-seq data have been deposited in the European Genome-phenome Archive database (accession numbers EGAD00001002323, EGAD00001002438, and EGAD00001002476).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Hélène Merle-Béral for fruitful discussions; Remi Brissiaud, Philippe Rameau, Marie-Claire Grison, Laurent Merlin, Myriam Boudjoghra, Claire Quiney, and Marc Le Lorch for technical assistance; Nadia Bougacha for preliminary assessments; and Stéphanie Mathis, Chantal Brouze, Justine Siavellis, Catherine Godon, Pierre Lemaire, Xavier Troussard, Valérie Bardet, Eric Jeandidier, Dominique Penther, Jean-Baptiste Gaillard, Vincent Leymarie, Annie Brion, Catherine Thieblemont, Cécile Tomowiak, David Ghez, Jean-Marc Zini, and Kamel Laribi for their contribution in the patient selection, the Groupe Francophone d’Hématologie Cellulaire.
This project was funded by the Institut Thématique Multi-Organisme Cancer, the Institut National du Cancer, GEFLUC, Association Laurette Fugain (ALF 14/08), and Roche Diagnostics. E.P. and L.J. received fellowships from the French Research Ministry and Société Française d’Hématologie and Fondation ARC, respectively. This project is part of the International Cancer Genome Consortium program. This work was supported by CURAMUS (Cancer United Research Associating Medicine, University and Society; INCA-DGOS-Inserm_12560).
Authorship
Contribution: F.N.-K., O.A.B, S.A.S., and E.C. designed the study; E.C., E.P., M.D., D.R.-W., C.D., C.G., M.Y., L.J., C.L., F.D., M.L.G.-T., N.D., P.D., S.B., L.W., and S. Scheinost performed experiments and analyzed data; K.M., C.S., L.B., and J.-F.L. performed the morphological review; C.A., V.L., J.G., V.E., B.G., E.C.-B., M.M., C.L., N.N., A.I., S. Struski, M.-A.C.-R., B.Q., S.F.-F., N.A., and I.R.-W. provided samples and clinical data; and E.C., F.D., T.Z., S.A.S., O.A.B. and F.N.-K. wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
A complete list of the members of the Groupe Francophone de Cytogénétique Hématologique (GFCH) and the French Innovative Leukemia Organization (FILO) appears in the supplemental appendix.
Correspondence: Florence Nguyen-Khac, Service d'Hématologie Biologique, Bâtiment Pharmacie, Pitié-Salpêtrière/Charles Foix University Hospital, 83 Bd de l'Hôpital, F-75013 Paris, France; e-mail: florence.nguyen-khac@aphp.fr.




![Overall survival in patients with B-PLL. Panels A-B as a function of MYC aberrations [t(MYC) or gain] and the presence or absence of MYC aberration; the t(MYC) and MYC gain data were pooled. (A) t(MYC) median (95% CI), 57.5 months (25.7-132.1); MYC gain median (95% CI), 66.5 months (4.7-undetermined). (B) MYC aberration [t(MYC) + MYC gain] median (95% CI), 57.5 (25.7-132.1). (C) MYC and del17p status. MYC aberration without del17p median (95% CI), 125.7 months (52.2-132.1); MYC aberration with del17p median (95% CI), 11.1 months (4.7-66.5). ab, aberration.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/134/21/10.1182_blood.2019001187/7/m_bloodbld2019001187f3.png?Expires=1765187031&Signature=nex7hfe~r1nfl4urvA1qT834TRWxw6w73PoSq6a4mes5KNHMpsFoUwsQZUeZeQ26NvhZgVEwzFiLO9RDU7q02qg7FE5Y9SudxjgNXPQ3EOxFnsb8PmZfcwUXJ~Ih9ZmebWbje795jVEdUKONkSEg9zijYgmoOdlDiaVSs2i7aYnkILCLvcZCgLWPHMettW3wTRBjzq~U1qhoPRCQg4nuXXu0GuBu3JOv3-146zkcoUTtO2jJOxdxhiLEzZjzHPcdwV-PbflcP~iMizS-8kcV1tAyGfg7VtYy2Wcw~XQUHakMZcjWgoBINDoBBcAF4XojxCpaFb0NjejbQaPi24mY5w__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
![The combination of drugs frequently used to treat B-cell malignancies with iBETs enhances the killing of primary B-PLL cells with t(MYC). (A) B-PLL cells from 3 patients with t(MYC) were treated with the indicated concentrations of ibrutinib, idelalisib, venetoclax, or fludarabine in the presence or absence of OTX015 (500 nM). Viability was assessed with an ATP-based CellTiter-Glo 2.0 kit. (B) B-PLL cell viability was measured and analyzed as in panel A in a patient with del17p but no MYC aberration. (C) Cell death was quantified in primary B-PLL cells from patient BPLL_8 [with t(MYC)] with or without pretreatment with OTX015 (500 nM) and exposure for 48 hours to ibrutinib (7.5 µM), idelalisib (50 µM), or venetoclax (10 nM). The percentages refer to Annexin-V–positive or Annexin-V–/propidium iodine–positive cells. (D) Primary B-PLL cells from BPLL_34 (with del17p but no MYC aberration) were treated with drugs and analyzed as in panel C. Bars represent the mean ± SEM.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/134/21/10.1182_blood.2019001187/7/m_bloodbld2019001187f4.png?Expires=1765187031&Signature=SYMnL1OOlm3qV3VLOZYNdMGP8ubatuTFeRGaHDn64n1GBOGKFK7jJjuXkRZy3TQYsbK-aE9Zlk-qU5ABXEDats7uXRlqLc~FcMJb2ma2~m1FzKbs11yn~823AOkonSFPfG2XP5oQhAoPCi97teapsZKraEwf4HL2BQV0urVoon5Gz7aAkLN-iU98yeiAi8BLpaGkFwgUNIdMNxmnk65WYTkMuRHcJrc04h7a~spCxoYRyWp19bS6RTtP~F5jy6ji7w2RKDDNM4GKC7sBBEOLLdUDpeUrytbanutYV550vTwLIdORL~Lr8p~MvQUB~WQMNGAa5rQjQJaL4vZI9ahDPg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
