

In this issue of Blood, 1 have conducted a comprehensive genetic, clinical, and drug sensitivity analysis of a large series of B-prolymphocytic leukemia (B-PLL) cases and illustrated important new findings with implications for predicting clinical outcome.
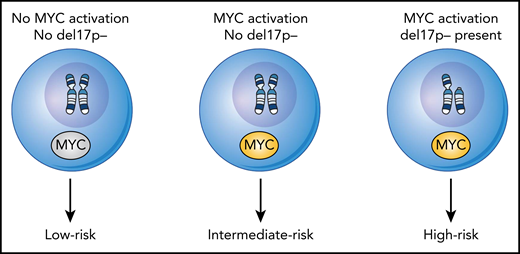
Model for the subsets of B-PLL based on their genetic parameters and linked to clinical outcome of overall survival. Using the presence or absence of MYC activation and del17p− status, 3 subsets of B-PLL can be discerned. Thus, for leukemic cells characterized with the following possibilities: with no MYC activation and no del17P− (ie, low risk), leukemic cells with MYC activation and no del17P− (ie, intermediate risk), and for leukemic cells with both MYC activation and del17P− (ie, high risk) there exists a hierarchy for overall survival.
Model for the subsets of B-PLL based on their genetic parameters and linked to clinical outcome of overall survival. Using the presence or absence of MYC activation and del17p− status, 3 subsets of B-PLL can be discerned. Thus, for leukemic cells characterized with the following possibilities: with no MYC activation and no del17P− (ie, low risk), leukemic cells with MYC activation and no del17P− (ie, intermediate risk), and for leukemic cells with both MYC activation and del17P− (ie, high risk) there exists a hierarchy for overall survival.
B-PLL being a recognized but rare chronic lymphoid leukemia for decades, there are still lively conversations among hematologists and hematopathologists when it comes to diagnosis and management. From a practical perspective, there has been a lack of large studies that help the practicing hematologist predict the clinical course for B-PLL and provide suitable and effective, modern treatment options for those patients.
Recognized by Galton and colleagues in 1974,2 B-PLL remains poorly understood.3,4 The diagnosis of B-PLL is based on finding a prominent number of circulating prolymphocytes. However, there is considerable morphologic and immunophenotypic overlap with other lymphoid disorders, including mantle cell lymphoma (MCL), chronic lymphocytic leukemia with increased prolymphocytes, and splenic marginal zone lymphoma (SMZL).5 Flow cytometry is a useful diagnostic tool, but there is no unique immunophenotype for B-PLL with bright expression of surface immunoglobulin M, clonal light chain, along with CD19, CD20, CD22, HLA-DR, CD79b, FMC7, and typically no CD23 or CD56 ; CD5 can be expressed in up to 20% to 30% and CD23 in 10% to 20% of B-PLL. The immunophenotype can overlap with other lymphoid leukemias.
B-PLL is also unique among the chronic lymphoid leukemias because it is not a diagnosis typically recognized via tissue biopsy. This has hampered our understanding of its basic biology, but also mandates the consideration of a tissue biopsy in these patients to better understand what disorder we are dealing with and to exclude the more common types of lymphoid malignancies. This includes MCL and also SMZL in the differential diagnosis because up to 20% of SMZL may be CD5-positive and can show p53 and MYC mutations later in the disease, similar to that seen in B-PLL. Often a consideration of the smear cytology or obtaining a tissue biopsy will provide a clear diagnosis.
Because of the inability to exclude MCL by morphology or by immunophenotype, genetic or immunohistochemical stains are necessary to exclude MCL when considering a B-PLL. Genetic studies describe abnormalities including 11q−, 13q−, 14q− anomalies, but have not yet provided a clear and distinct genetic profile for B-PLL. Other notable genetic findings in B-PLL include a trend to mutated IGVH status, >50% detection of p53 mutations, and identification of recurrent C-MYC abnormalities.7-9
To all these conundrums swirling around B-PLL, there is no agreed upon prognostic model to use in counseling B-PLL patients with the exception that p53 mutations confer a worse outcome, and therapies are not highly effective in the latter cohort. However, p53 mutated B-PLL may be more responsive to B-cell receptor–directed therapies given recent reports.10
The study by Chapiro et al attempted to answer some of these questions as they amassed a relatively large series of B-PLL. These patients were carefully assessed retrospectively for B-PLL by using morphologic evaluation with a panel of experienced hematopathologists. However, similar to other studies of B-PLL, no lymph node, spleen, or other tissue biopsies were provided to confirm the histopathologic diagnoses, and thus overlap with other lymphoid malignancies cannot be totally excluded. Importantly, there was an extended follow-up time for these cases allowing for overall survival to be determined.
The investigators had considerable molecular information at initial diagnosis on 21 of the 34 cases that included cytogenetics, fluorescence in situ hybridization, whole-exome sequencing, targeted deep sequencing, RNA expression profiling, and IGVH mutation analysis. Specifically, notable findings were that C-MYC abnormalities of either translocations or gains are present in >75%, adding more evidence of its involvement in B-PLL, a high incidence of complex karyotypes, and a high frequency of p53 mutations. Although in large part confirmatory findings, these data add significantly to the characterization and potential critical molecular mechanisms extant in B-PLL. Other notable genetic findings in this paper included the detection of mutations in a putative tumor suppressor gene, BCOR, along with MYC translocations that may be unique to B-PLL; a majority of cases had mutated IGVH status, and not unexpectedly a high frequency of 17p− patients. In respect to pathogenesis of B-PLL, they also found clustering in some MYC translocation cases with the presence of clonal mutations in MYC, BCOR, and SF3B1 genes.
From the composite of the genetic data, the authors propose a novel hierarchical model for 3 cohorts based on overall survival; a highest-risk group that features MYC activation (presence of translocation or gains) along with 17p−, an intermediate-risk group with MYC activation only, and an indolent, low-risk group without MYC activation (see figure).
In selected cases, the authors conducted preclinical work with a combination of novel agents (Ibrutinib, Venetoclax, Idelalisib) and 2 bromodomain inhibitor drugs, OTX015/JQ1, and describe potent killing activity with these combinations in patients with an MYC translocation. Given the epigenetic regulation capacity of OTX015, the authors speculate that this drug class may work by suppressing MYC-enhanced genes.
This is in total a well-done study in an extremely rare disease, but questions arise out of their main conclusions. How will future studies affirm the hierarchical model and their survival outcomes of B-PLL given its rarity and difficulty in establishment of specific diagnoses? Can the detection of MYC activation and its apparent vulnerability be truly exploited in the treatment of B-PLL? What are we to make of the preclinical data showing that B-cell signaling agents along with bromodomain inhibitors are effective in the reduction in B-PLL viability for some of the higher-risk cohort? One issue is that the B-PLL cells used were likely not freshly harvested and thus perhaps more likely to succumb to in vitro drug exposures. Also, is there a gold-standard preclinical killing assay that should be used in these experiments: ATP-based or Annexin PI flow-based approaches?
Despite these caveats, the investigators are to be applauded for adding significantly new, provocative biologic and clinical information in B-PLL.
Conflict-of-interest disclosure: The authors declare no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal