

Unexplained weight loss and cachexia are key hallmarks of cancer and advanced myeloproliferative neoplasms (MPNs). In this issue of Blood, describe their investigation of the metabolic response to JAK2 mutation in mouse models, which demonstrated that the presence of the JAK2-mutant clone leads to hypoglycemia and adipose tissue atrophy as a result of scavenging nutrients to meet the increased glucose requirements of erythroid precursors.1 They identify the enzyme Pfkfb3 as a key mediator of glycolysis and show that inhibition of Pfkfb3 reverses hypoglycemia and reduces the hematopoietic manifestations of the disease. These data offer a first mechanistic insight into a poorly characterized aspect of MPN disease biology, and provide a clear preclinical rationale for further investigation of the metabolome as a therapeutic target.
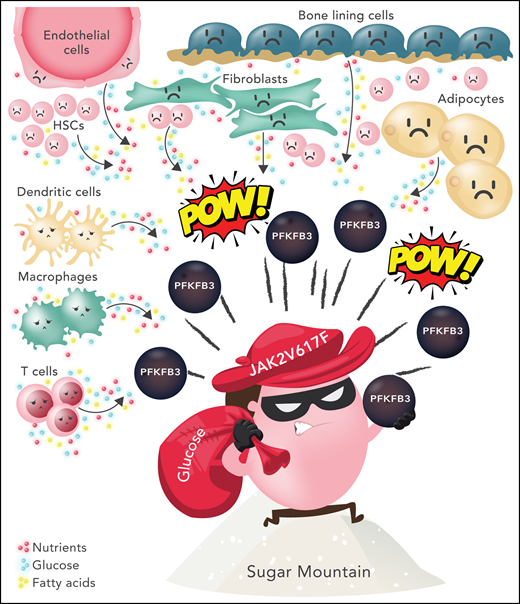
Glucose subversion by JAK2 V617F-positive MPNs. JAK2 V617F-positive cells exist within a tumor microenvironment consisting of multiple cell types, including fibroblasts, endothelial cells, bone lining cells, adipocytes, and neighboring non-mutant hematopoietic stem and progenitor cells. Surrounding immune cells (eg, dendritic cells, macrophages, and T cells) are thought to be overall immunosuppressive. Each cell type has unique metabolic demands. In this cartoon, an MPN cell is depicted using Pfkfb3, a rate-limiting enzyme in glycolysis, to increase glycolysis and usurp nutrients (glucose, fatty acids, amino acids) from the microenviroment. It is assumed that the relative glucose deprivation of surrounding cells leads to a competitive advantage for the MPN clone. HSC, hematopoietic stem/progenitor cells. Professional illustration by Somersault18:24.
Glucose subversion by JAK2 V617F-positive MPNs. JAK2 V617F-positive cells exist within a tumor microenvironment consisting of multiple cell types, including fibroblasts, endothelial cells, bone lining cells, adipocytes, and neighboring non-mutant hematopoietic stem and progenitor cells. Surrounding immune cells (eg, dendritic cells, macrophages, and T cells) are thought to be overall immunosuppressive. Each cell type has unique metabolic demands. In this cartoon, an MPN cell is depicted using Pfkfb3, a rate-limiting enzyme in glycolysis, to increase glycolysis and usurp nutrients (glucose, fatty acids, amino acids) from the microenviroment. It is assumed that the relative glucose deprivation of surrounding cells leads to a competitive advantage for the MPN clone. HSC, hematopoietic stem/progenitor cells. Professional illustration by Somersault18:24.
A major reprogramming of cellular energy metabolism has been described as a core cancer-associated trait.2 Tumor microenvironments have limited glucose available, and cancer cells compete for nutrients with surrounding stromal cells and the immune compartment. This leads to increased glucose uptake and consequent nutrient deprivation for the surrounding tumor niche (see figure), potentially hampering immune surveillance and reprogramming the entire tumor niche in favor of cancer cell survival and proliferation. The “Warburg effect,” first described around 100 years ago, describes how proliferating tumor cells reprogram their energy metabolism in favor of glycolysis and away from mitochondrial aerobic respiration, even in the presence of plentiful oxygen (aerobic glycolysis).2 New evidence in colorectal cancer indicates that the loss of glycolytic regulation may be a key targetable step during tumorigenesis.3 Although the cellular components of the tumor microenvironment have been well studied, insights on how metabolites in the tumor niche are subverted by the malignant clone are lagging behind. In parallel, there is increasing awareness from a public health perspective that obese populations and consumption of a high sugar diet may increase the risk of certain types of tumors. Furthermore, although there is limited clinical evidence of efficacy, there is increasing preclinical rationale for dietary or drug-induced glucose restriction as a mechanism to inhibit tumor growth.4,5
In hematologic malignancies, there is evidence that acute myeloid leukemia cells usurp glucose by inducing insulin resistance and lipolysis from adipose tissue,6 and that leukemia-initiating stem cells are more sensitive to inhibition of glycolysis than normal stem cells.7 Hypoglycemia in patients with myeloid malignancy has also been described, although it is considered artifactual because of in vitro glycolysis in the blood sample tube.8
In this study, mouse models of JAK2 V617F and JAK exon 12–mutated polycythemia vera (PV) develop hypoglycemia and cachexia with adipose tissue loss despite increased food intake. The phenotype is not corrected but rather worsened by a high glucose diet, which seems to fuel erythrocytosis and splenomegaly. In contrast, limiting glucose supply by intermittent fasting ameliorates MPN features. A strong inverse correlation was observed between blood glucose and hemoglobin, reflecting the increased glucose consumption by cycling erythroid cells. Pfkfb3, a rate-limiting enzyme in glycolysis, was significantly increased in mutant hematopoietic stem and progenitor cells, and inhibition with the small molecule 3PO dramatically reduced myeloproliferation in vivo and induced a disease response in mouse models that was additive to ruxolitinib. There was no correlation between blood glucose and platelet, neutrophil, or monocyte counts. Hypoglycemia was not observed in a JAK2-mutant essential thrombocythemia (ET) model, but did occur in a model of erythropoietin-induced erythrocytosis in the absence of a JAK2 V617F clone. 3PO did not reduce the JAK2 V617F allele burden, providing further evidence that the observed glycolytic dependency may not be JAK2 V617F dependent but may be secondary to the increased erythropoiesis seen in this disease model.
One outstanding question relates to how these observations translate to human myeloproliferative disorders and more broadly to cancer-induced cachexia. Cachexia affects 50% to 80% of all cancer patients, shortens survival, is a major cause of impaired quality of life, and remains poorly characterized.9 Cachexia and hypermetabolism are cardinal features of myelofibrosis (MF) but have historically been attributed to early satiety secondary to splenomegaly, and increased inflammatory cytokines. Patients with MF do not however have increased erythropoiesis, whereas in PV, in which erythrocytosis predominates, weight loss is not a typical feature. Future insights from studies at single-cell resolution may reveal substantial metabolic heterogeneity among tumor cells and be able to properly dissect differences in metabolic state between malignant cells, infiltrating immune cells, and stromal cells that may be masked by studies of cells sampled in bulk.10
There is a major unmet need for novel and effective therapies for MPNs, particularly for patients with MF and patients with PV or ET who do not respond to first-line therapies. Targeting the MPN metabolome represents an exciting and innovative approach. Although Pfkfb3 requires further formal validation in patients before it can be confirmed as a tractable target in MPN, derivatives of 3PO are already in clinical trials of solid cancers, and dietary recommendations are under investigation. Understanding the cellular and molecular mechanisms for cachexia and how normal metabolic processes are hijacked by tumor cells to support unlimited cellular proliferation has great potential for novel and rapidly translatable insights into cancer pathobiology.
Conflict-of-interest disclosure: The authors declare no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal