

In this issue of Blood, used genetic and pharmacologic approaches to demonstrate a role of eosinophils in mouse models of atherosclerosis and arterial thrombosis.1 In addition, analysis of thrombi from patients with acute myocardial infarction or stent thrombosis revealed that the highest numbers of eosinophils in stent thrombi are found in female patients.
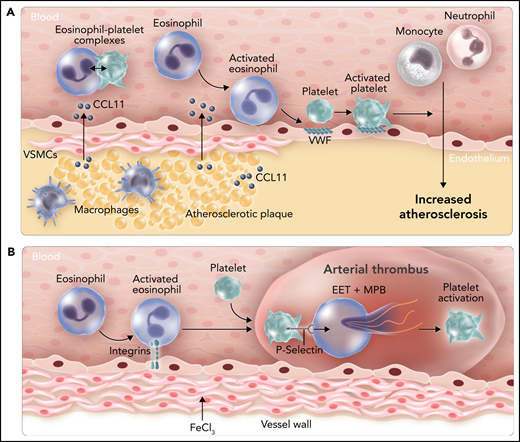
Roles of eosinophils in atherosclerosis and arterial thrombosis in mice. (A) Expression of the chemoattractant CCL11 by cells within atherosclerotic plaques leads to the recruitment of eosinophils. Binding of eosinophils to the endothelium leads to surface expression of von Willebrand factor (VWF) and binding and activation of platelets that enhance leukocyte migration into the vessel wall. CCL11 also activates eosinophils and increases levels of circulating eosinophil-platelet aggregates. (B) Administration of FeCl3 to the outside of the carotid artery leads to integrin-dependent binding of eosinophils. P-selectin–dependent platelet binding to eosinophils induces the release of EETs and MBP, which enhance platelet activation. VSMC, vascular smooth muscle cell. Professional illustration by Somersault18:24.
Roles of eosinophils in atherosclerosis and arterial thrombosis in mice. (A) Expression of the chemoattractant CCL11 by cells within atherosclerotic plaques leads to the recruitment of eosinophils. Binding of eosinophils to the endothelium leads to surface expression of von Willebrand factor (VWF) and binding and activation of platelets that enhance leukocyte migration into the vessel wall. CCL11 also activates eosinophils and increases levels of circulating eosinophil-platelet aggregates. (B) Administration of FeCl3 to the outside of the carotid artery leads to integrin-dependent binding of eosinophils. P-selectin–dependent platelet binding to eosinophils induces the release of EETs and MBP, which enhance platelet activation. VSMC, vascular smooth muscle cell. Professional illustration by Somersault18:24.
Eosinophils are a well-known part of the immune system used to fight infections and parasites in vertebrates. Activated eosinophils release several cationic proteins, such as major basic protein (MBP), eosinophil cationic protein (ECP), and eosinophil peroxidase (EPX), as well as DNA traps (called eosinophil extracellular traps [EETs]) that are designed to kill pathogens. However, eosinophilic toxins and EETs also cause cellular damage. For instance, high levels of eosinophils lead to inflammation and tissue damage in patients with eosinophilic asthma.
Marx et al first analyzed the role of eosinophils in atherosclerosis by using an eosinophil-deficient mouse line called ΔdblGATA1.2 These mice have a deletion of the GATA1 binding site in the GATA1 promoter that leads to a selective deficiency of eosinophils. ΔdblGATA1 mice were crossed with the atherosclerosis-prone mouse line ApoE−/−, and double knockout mice and controls were fed a cholesterol-rich western diet for 13 weeks. Atherosclerosis was significantly reduced in the aortic arch and the aortic root in ApoE−/−;ΔdblGATA1 mice compared with ApoE−/− mice. These lesions also had reduced percentages of macrophages, neutrophils, and smooth muscle cells. Importantly, the eosinophil chemoattractant protein CCL11 was expressed within atherosclerotic lesions and released into the bloodstream, which is likely responsible for the recruitment and activation of eosinophils. Binding of activated eosinophils to the endothelium increased platelet recruitment by inducing the exposure of von Willebrand factor. Importantly, platelets have been shown to promote atherosclerosis.3 Taken together, these results suggest that eosinophils enhance atherosclerosis in mice by activating the endothelium and increasing platelet recruitment (see figure panel A). A complementary approach to using ΔdblGATA1 mice would have been to determine the effect of depleting and inhibiting eosinophils in wild-type mice using an anti-Siglec-F antibody.
In the second part of their study, Marx et al investigated the role of eosinophils in arterial thrombosis by using an FeCl3 carotid artery mouse model. The effect of eosinophil deficiency in this model was quite subtle because it did not affect the time of induction of thrombus formation and arterial occlusion, but it did reduce thrombus stability. It would be interesting to know whether neutrophils have a similar effect on thrombus stability in this model. Deletion of the integrin regulatory protein Kindlin-3 in eosinophils also produced a shorter duration of occlusion similar to that in mice lacking eosinophils, which indicated a role for integrins in the recruitment of eosinophils to the damaged endothelium. Platelets have been shown to activate eosinophils, and in this study, platelet binding to eosinophils caused a calcium burst inside the cells.
Neutrophil extracellular traps (NETs) have recently been shown to be present in both arterial and venous thrombi.4 NETs enhance thrombosis in a variety of ways, such as recruiting platelets and stabilizing thrombi.4 More recently, other cell types, including eosinophils, have been shown to release DNA traps.4 Nicotinamide adenine dinucleotide phosphate oxidase was shown to be required for the release of EETs.5 Marx et al distinguished EETs from NETs by colocalizing extracellular DNA with the eosinophil-specific MBP. They found that 27% of extracellular traps in the thrombi originated from eosinophils. However, it is possible that granular proteins released from eosinophils can also bind to NETs and a positive control for the binding of a neutrophil protein to extracellular DNA was not used. As has been shown with neutrophils,6 platelets induced EET formation in a P-selectin–dependent manner.1 Interestingly, mice deficient in the eosinophil granular protein MBP, but not in EPX, exhibited decreased thrombus stability. Finally, inhibition of eosinophils with an anti-Siglec-F antibody also reduced thrombus stability. These studies demonstrate a role of eosinophils in arterial thrombosis in the FeCl3 mouse model (see figure panel B). Further studies could investigate the relative contribution of EET formation vs other eosinophil-dependent mechanisms in thrombus formation, such as release of elastase.
The studies by Marx et al are consistent with a recent study showing that eosinophil-deficient mice and wild-type mice treated with an anti-Siglec-F antibody had reduced thrombosis in an FeCl3 inferior vena cava mouse model.7 However, it is a concern that Marx et al used only a single model of arterial thrombosis—the FeCl3 model—which uses a non-physiologic agent to injure the vessel wall.8 Importantly, unlike neutrophils and NETs,9 an absence of eosinophils did not reduce venous thrombosis in an inferior vena cava flow restriction mouse model.7 Furthermore, the atherosclerosis and thrombosis parts of the study by Marx et al could have been tied together nicely if they had determined the role of eosinophils in atherothrombosis. Atherosclerotic plaques in mice do not spontaneously rupture, but they can be induced to rupture by ultrasound.10 Therefore, it would be relatively easy to compare carotid artery thrombosis in ApoE−/− mice with or without inhibition of eosinophils.
How do these mouse studies translate to the clinic? A recent epidemiology study found that the eosinophil granular protein ECP was associated with advanced atherosclerosis and thrombotic events.7 Marx et al measured the number of eosinophils in arterial thrombi from patients with acute myocardial infarction and stent thrombosis. They observed significantly higher numbers of eosinophils in stent thrombi from female patients compared with male patients. The authors proposed that this subgroup of patients might benefit from an eosinophil-inhibiting strategy. However, it seems premature to suggest that inhibiting eosinophils represents a novel way to safely reduce stent thrombosis without more data on the role of eosinophils in thrombosis in humans.
Conflict-of-interest disclosure: The author declares no competing financial interests.

