

Key Points
Eosinophils promote atherosclerotic plaque formation and thrombosis.
Eosinophils form extracellular traps triggered by platelets, which enhance thrombus stability through major basic protein.

Abstract
Clinical observations implicate a role of eosinophils in cardiovascular diseases because markers of eosinophil activation are elevated in atherosclerosis and thrombosis. However, their contribution to atherosclerotic plaque formation and arterial thrombosis remains unclear. In these settings, we investigated how eosinophils are recruited and activated through an interplay with platelets. Here, we provide evidence for a central importance of eosinophil-platelet interactions in atherosclerosis and thrombosis. We show that eosinophils support atherosclerotic plaque formation involving enhanced von Willebrand factor exposure on endothelial cells and augmented platelet adhesion. During arterial thrombosis, eosinophils are quickly recruited in an integrin-dependent manner and engage in interactions with platelets leading to eosinophil activation as we show by intravital calcium imaging. These direct interactions induce the formation of eosinophil extracellular traps (EETs), which are present in human thrombi and constitute a substantial part of extracellular traps in murine thrombi. EETs are decorated with the granule protein major basic protein, which causes platelet activation by eosinophils. Consequently, targeting of EETs diminished thrombus formation in vivo, which identifies this approach as a novel antithrombotic concept. Finally, in our clinical analysis of coronary artery thrombi, we identified female patients with stent thrombosis as the population that might derive the greatest benefit from an eosinophil-inhibiting strategy. In summary, eosinophils contribute to atherosclerotic plaque formation and thrombosis through an interplay with platelets, resulting in mutual activation. Therefore, eosinophils are a promising new target in the prevention and therapy of atherosclerosis and thrombosis.
Introduction
Inflammation is increasingly recognized as a central driver of atherosclerosis and more recently also of thrombosis, but the cellular and molecular mechanisms are still incompletely understood. The need for new therapeutic targets in this field is supported by the fact that cardiovascular diseases still represent the leading cause of death worldwide.1 In the vast majority of patients, the pathophysiologic basis for these diseases is atherosclerosis. Canonically, the innate immune system is considered as the key driver in the development of atherosclerotic plaques, and anti-inflammatory therapies are currently evolving.2-4 Plaque rupture results in the exposure of strongly procoagulant subendothelial matrix, including tissue factor (TF), and the subsequent recruitment and activation of platelets and leukocytes leads to life-threatening arterial thrombosis.5-7 In the last few years, the impact of immune cells has moved into focus with respect to their involvement in thrombosis, and multiple studies have proved that neutrophils and monocytes contribute to the pathogenesis of atherosclerosis and thrombosis.8-10 Activated platelets and endothelial cells recruit these immune cells, which, in the event of plaque rupture, promote thrombus formation and stabilization by TF delivery and formation of neutrophil extracellular traps (NETs).11,12
Several clinical observations suggest that, besides these classical players, eosinophils may also play a role in atherosclerosis and arterial thrombosis. Several lines of evidence link eosinophils to cardiovascular events, but it is not known which specific aspect of the pathophysiology they contribute and whether they interact with platelets as key drivers of these processes. In atherosclerosis, elevated blood levels of the eosinophil cytotoxic effector protein eosinophil cationic protein (ECP) correlate with the severity of arterial stenosis and predict atherosclerotic burden.13,14 Eosinophils were absent in histologic examinations of stable human atherosclerotic plaques, but they were detected in ruptured plaques. In addition, the potent eosinophil chemoattractant and activator CCL11 (eotaxin-1) is overexpressed in atherosclerotic lesions.15-17 These findings indicate that activated eosinophils may play a role in atherosclerosis, possibly involving activation and shedding of their cytotoxic granules. Eosinophil granules contain the cationic proteins major basic protein (MBP), eosinophil peroxidase (EPX), ECP, and eosinophil neurotoxin, which cause tissue damage and inflammation.18 Eosinophils are also associated with arterial thrombosis in several clinical studies. We detected eosinophil accumulation in thrombi retrieved from patients presenting with myocardial infarction.19 Moreover, high eosinophil counts in thrombi are linked to increased thrombus size.20 In line with this, a genome-wide association study for sequence variants affecting systemic eosinophil counts found an association with myocardial infarction.21 Recently, we and others found a procoagulant effect of eosinophils by delivering activated TF in a murine model of venous thrombosis, but whether there is an activating interplay between platelets that enhance arterial thrombosis and the formation of plaque in the setting of atherosclerosis remains unclear.22,23
Here, we provide evidence that eosinophils enhance atherosclerotic plaque formation as well as thrombosis through interaction with platelets, which results in mutual activation. Eosinophils are stimulated by CCL11 in atherosclerosis, which is associated with endothelial activation and exposure of von Willebrand factor (VWF). This promotes atherogenic platelet adhesion to the vessel wall. In response to endothelial injury, eosinophils are rapidly recruited to the injury site in an integrin-dependent manner and get activated by direct interactions with platelets. Eosinophils in turn promote thrombus development by forming eosinophil extracellular traps (EETs), which contain MBP and thereby reinforce platelet activation. In a translational approach, we demonstrate that pharmacologic inhibition of EET formation decreases thrombus stability. This mechanism might be particularly important in female patients with stent thrombosis (ST), who show the highest eosinophil numbers within coronary artery thrombi. In summary, we reveal an interplay of eosinophils and platelets as a new mechanistic principle enhancing atherosclerosis and thrombosis.
Methods
Animals
Specific-pathogen–free C57BL6/J mice were purchased from Charles River. Kindlin-3fl/fl and PC-G5-tdTfl/fl mice were cross-bred with EoCrecre mice. To investigate the importance of integrins for eosinophil recruitment in arterial thrombosis, we used EoCre;Kindlin-3fl/fl mice, in which eosinophils lack the pan-integrin regulatory protein Kindlin-3. In EoCrecre;PC-G5-tdTfl/fl mice, eosinophils express a constitutive tdTomato signal and a green fluorescent protein signal coupled to a calcium indicator that allows in vivo assessment of calcium levels in eosinophils.24-26 EPX−/− and MBP−/− mice were provided by James Lee, generated as described, and compared with littermate controls. These mouse lines lack the cytotoxic MBP and EPX, respectively.27,28 To determine the role of an eosinophil-inhibiting antibody, C57BL6/J mice received a monoclonal rat anti-mouse Siglec-F antibody (BD Pharmingen) intravenously. Mice age 8 to 14 weeks and age- and sex-matched were included in the experiments. Surgical interventions were performed under anesthesia using midazolam (5 mg/kg), fentanyl (0.05 mg/kg), and medetomidine hydrochloride (0.5 mg/kg). All animal studies were approved by local legislation for protection of research animals (Regierung von Oberbayern).
Evaluation of lesion size and composition
ApoE−/− mice were bred with eosinophil-deficient ΔdblGATA1 mice backcrossed to a C57Bl6 background for at least 10 generations.29 After weaning, mice were fed a high-cholesterol diet for 13 weeks that contained 22% fat and 0.2% cholesterol (Sniff EF D12079). ApoE−/−;ΔdblGATA1−/− mice were compared with ApoE−/−;ΔdblGATA1+/+ littermate controls. To determine lesion size relative to the aortic root, Oil-Red-O staining was used. Immunofluorescence stainings were performed in 10 sequential sections per mouse using an anti-Siglec-F (BD Pharmingen), anti-VWF (DAKO), anti-myeloperoxidase (A0398; DAKO), anti-LGAL-S3 (CL8942AP; Cedarlane), and anti-ACTA-2 antibodies (F3777; Sigma). Data for neutrophil, macrophage, and smooth muscle cell accumulation are given as area of the specific staining relative to plaque area. Images were taken with an epifluorescence microscope (20× magnification) and analyzed with Fiji software. In addition, the lipid areas in the aorta were stained with Sudan stain in a whole-mount staining procedure. Images of the aorta were taken with 1.6× magnification using a brightfield microscope, and the lipid area was analyzed with Fiji software.
ELISA
Plasma concentrations of activated protein C (APC) as well as concentrations of total cholesterol were determined using enzyme-linked immunosorbent assay (ELISA) kits (Quantikine ELISA Kit, R&D Systems) and Cholesterol Assay Kit (Abcam). Interleukin-5 (IL-5), CCL11, and VWF were quantified by using ELISA kits.
Assessment of platelet activity
Using heparin coated syringes, whole blood was drawn from ApoE−/−;ΔdblGATA1 mice. Platelet-rich-plasma was isolated, 5 µL adenosine 5′-diphosphate (ADP) (20 mM in 100 µL phosphate-buffered saline [PBS]; Sigma) was added, and platelet aggregation and adenosine triphosphate release were measured using an aggregometer, including measurement of luminescence (Chrono-Log 490-2D). Blood was drawn from MBP−/− and wild-type mice by using syringes coated with heparin and mixed with 500 µL Tyrodes buffer (pH 7.4). After adding IL-5 (10 µg/mL; R&D Systems) followed by CCL11 (10 µg/mL; R&D Systems), ADP (20 mM in 100 µL PBS; Sigma) was added, and platelet aggregation was measured (optical Lumi-Aggregometer model 700-4). Platelets from human blood were incubated with supernatant from stimulated (50 ng/mL IL-5) or resting eosinophils (6 × 105 cells per mL) and medium containing IL-5 (50 ng/mL). Exposure of VWF on platelets was assessed by fluorescence-activated cell sorting (FACS) using an anti-VWF antibody (GeneTex).
Mouse model of arterial thrombosis
Mice were anesthetized, reagents were injected into the tail vein, and the left carotid arteries were exposed by a paramedian incision. To induce thrombus formation, a Whatman paper (2 × 2 µm; Sigma Aldrich) presoaked with 10% FeCl3 solution was placed beside the artery and removed after 3 minutes.30
Mouse model of mesenteric vessel thrombosis
After anesthesia, the cranial mesenteric artery and its branches were laid on a glass slide and injured by applying FeCl3. Thrombus formation was imaged by using a Zeiss Laser scanning microscope (LSM 880) with an airyscan module (40× magnification).
Intravital epifluorescence microscopy
To evaluate in vivo thrombus formation, murine platelets were labeled with 0.25 µL glycoprotein Ib (GPIb) antibody (Dylight488 anti-GPIβ; Emfret) per gram body weight. Imaging was performed with an Olympus BX51WI microscope using a 5× (NA 0.10) objective or a 20× (NA 0.95) water-immersion objective and an ORCA-ER charge coupled device camera (Hamamatsu). Thrombi were defined as occlusive when no continuous flow of blood was detectable. Eosinophils were defined as adherent when they stayed at the same position for >3.3 seconds. The GPIb antibody does not allow investigations on a single-cell level; hence, platelets of ApoE−/−;ΔdblGATA1−/− mice and littermate controls were isolated and stained with 5 µMol highly fluorescent 2′,7′-dichlorofluorescein (DCF) (Invitrogen). The number of platelets was determined, and each genotype-matched recipient received 10 µL per gram of body weight of the platelet suspension in a concentration of 1.5 × 105 cells per µL. An adhesion was counted as interaction when the platelets remained at the same position for >330 ms. For in vivo evaluation of the calcium signal in eosinophils after interaction with platelets, platelets needed to be stained with 5 µMol CellTracker Violet (Invitrogen) for visualization. Mice were imaged by using a Leica DM 6FS fluorescent microscope with a Zyla scientific complementary metal-oxide-semiconductor camera (Andor). Thrombus formation was observed in vivo for 45 minutes. Differential blood cell counts in mouse blood were analyzed by using an Idexx Procyte Dx hematology analyzer.
Immunofluorescence examination of arterial thrombi
Cryosections (30 sections per thrombus) were fixed with paraformaldehyde and stained with a rat anti-mouse Siglec-F antibody and rabbit anti-mouse MBP antibody (Abcam). Eosinophils in human thrombi were stained with a rabbit anti-human MBP antibody (Merck). Images were acquired using a Zeiss Imager M2 Axio epifluorescence microscope and processed using AxioVision SE64 Rel. 4.9 software. For quantification of EETs, confocal images were acquired with a Zeiss Laser scanning microscope (LSM 880) with an airyscan module (63× oil immersion lens). For quantification of EET formation, 3 distinct parameters had to be met: (1) extracellular DNA protrusions had to be present, (2) the protrusion had to originate from cells that stained positive for MBP, and (3) the structures had to be decorated with MBP. Eosinophils, neutrophils, NETs, and EETs were assessed by staining 5 sections each from the proximal, medial, and distal part of thrombi of ApoE−/− mice for 4′,6-diamidino-2-phenylindole (DNA), myeloperoxidase (Abcam), and citHistone3 (NETs; Abcam), as well as MBP (EETs). Images were analyzed by using Fiji software.
Isolation of human eosinophils
Blood was drawn from healthy, nonatopic donors in syringes containing 0.1 M citrate, followed by Ficoll gradient (GE Healthcare) centrifugation. Erythrocytes were lysed, and all of the following steps were performed at 4°C. Eosinophil isolation was performed by negative immunomagnetic selection, as previously described.31 Cells were pelleted by centrifugation for 10 minutes at 300g. Purity of isolation was verified by FACS for Siglec-8 and was confirmed to be >98%; there was no contamination with platelets as assessed by FACS for CD41.
Isolation of human platelets
Blood was drawn into citrate-containing syringes and diluted with Tyrodes buffer (pH 6.5). To avoid platelet aggregation during isolation, 10 µM prostacyclin (Santa Cruz Biotechnology Inc.) was added. The platelet-rich supernatant was collected and diluted with Tyrodes buffer (pH 6.5), and cells were pelleted by a second centrifugation step for 10 minutes at 120g. Then, 6 × 108 cells were resuspended in 1 mL Tyrodes buffer (pH 6.5), and 300 eosinophils per µL were incubated with 300 000 platelets per µL at 37°C for 45 minutes. Platelets were either activated by 0.01 units/mL thrombin (Sigma Aldrich) or left inactive. Eosinophils were incubated with a supernatant of platelets.
Analysis of eosinophil-platelet interaction
To assess EET formation, an anti-CD41 antibody (Abcam) and an anti-MBP antibody (Merck) were used. Nuclei and EETs were stained with a Hoechst stain (ThermoFisher). The percentage of EETs relative to total eosinophil count was investigated by taking images of 10 × 10 different fields. A Zeiss laser scanning microscope (LSM 880) with an airyscan module was used to evaluate MBP-platelet interactions.
FACS
EDTA-anticoagulated mouse whole blood was incubated with antibodies or respective isotype controls (1:50) for 15 minutes at room temperature, and red blood cells were lysed with FACS lysing solution (Cat No. 349202, BD Biosciences) before analysis. All samples were analyzed using a FACSCanto II or FACS Fortessa flow cytometer (BD Biosciences). Platelet eosinophil aggregates were measured using anti-CD41 pacific blue (clone MWReg30; eBioscience) and anti-Siglec-F APC-C7 (BD Pharmingen). Exposure of VWF and GPIb on platelets was assessed by using an anti-GPIb-X649 (clone Xia.G5; Cat No. M040-3, Emfret) and an anti-VWF fluorescein isothiocyanate (Cat No. GTX28822; Gentex) antibody.
Evaluation of VWF exposure by endothelial cells
Isolated human eosinophils (6 × 105 cells per mL) were activated with IL-5 (10 µg/mL) and CCL11 (10 µg/mL; R&D Systems). Human umbilical vein endothelial cells were cultivated in endothelial cell growth medium (Promega) until confluence, and 3 × 104 cells were stimulated with tumor necrosis factor α (100 ng/mL; Sigma Aldrich) as positive control, activated or resting eosinophil supernatant, or PBS containing IL-5 and CCL11 for 5 hours as vehicle control. Then, cells were fixed and stained with an anti-VWF antibody (2 µg/mL; Abcam). Images were taken with the oil immersion objective (60× magnification) of a Zeiss Laser scanning microscope (LSM 880) with an airyscan module.
Patients and retrieval of human thrombi
We examined thrombi from 282 patients in the PRESTIGE project who were suffering from a coronary ST.32 Patients with acute myocardial infarction (AMI) were included when thrombotic occlusion of a non-stented coronary artery was detected with an indication for removing the thrombotic material (Pronto thrombectomy catheter). Recruitment of these 282 patients occurred at a single center (Deutsches Herzzentrum München, Munich, Germany). Approval for this study was obtained from the local institutional review board (DHZ project No. 4007/11a; LMU project No. 18-343), and written informed consent was obtained in accordance with the Declaration of Helsinki.
Histology of human coronary artery thrombi
Thrombus specimens processed as previously described32 and tissue sections were stained with Luna (EMS).33 Specifically, serial cross sections 5 µm thick were cut. Paraffin-embedded tissue sections were deparaffinized by immersion in xylene and rehydrated in decreasing concentrations of ethanol. To remove all alcohol, slides were washed in warm tap water. After that, slides were placed into hematoxylin-Biebrich Scarlet Solution for 5 minutes. Sections were quickly dipped into 1% acid alcohol solution 3 to 8 times and then quickly rinsed in 3 changes of deionized water. In the next step, slides were quickly dipped 2 to 5 times in an ammonia and water solution until sections turned light blue (background), darker blue (nucleus), or bright red (eosinophil granules). Finally, sections were dipped 2 to 5 times in warm water, dehydrated by being passed through graded alcohols, and mounted with colorless mounting medium (Pertex). Images were acquired by using either a Zeiss Imager M2 Axio epifluorescence microscope and processed with AxioVision SE64 Rel. 4.9 software or a Leica DMRB epifluorescence microscope equipped with a Zeiss AxioCam and processed using AxioVision 4.6 software (Zeiss). Eosinophils were counted manually.
Statistical analysis
In Figure 1, data from human samples are shown as mean ± standard error of mean or mean ± standard deviation unless otherwise indicated. Statistical analyses were carried out using GraphPad Prism 7.04 and 8.0, and data were tested for normal distribution using the Kolmogorov-Smirnov test. The Student t test and Mann-Whitney U test, respectively, were performed to compare 2 groups. More than 2 groups were compared by using analysis of variance followed by the Tukey post hoc test. Logistic regression analysis was performed using R version 3.6.1. P < .05 was considered significant.
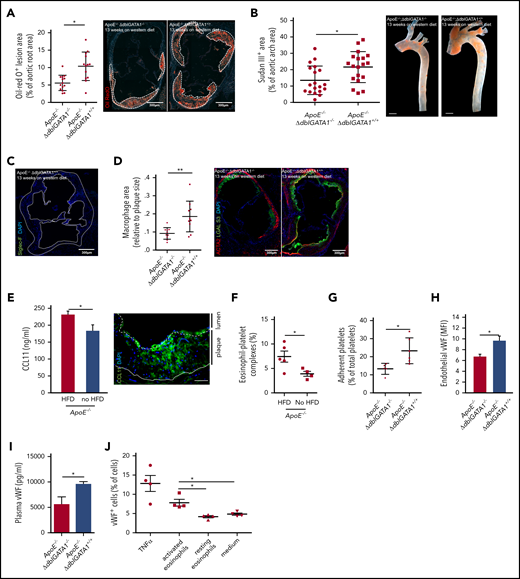
Eosinophils promote atherosclerotic plaque formation. (A) Atherosclerotic lesion formation (white dotted lines) assessed by lipid staining with Oil-Red O in the aortic root of eosinophil-deficient ApoE−/−;ΔdblGATA1−/− mice (n = 14: 7 female, 7 male) compared with littermate controls (ApoE−/−;ΔdblGATA+/+ (n = 13: 6 female, 7 male) after 13 weeks on an HFD. (B) Quantification of macroscopic lesion formation by Sudan III staining in the aortic arch of ApoE−/−;ΔdblGATA1−/− mice (n = 19: 9 female, 10 male) compared with ApoE−/−;ΔdblGATA+/+ littermate controls (n = 19: 9 female, 10 male). Scale bar, 200 µm. (C) Eosinophils were absent in plaques (white dotted line) of ApoE−/−;ΔdblGATA+/+ mice as assessed by staining for Siglec-F (green), nuclei are stained with 4′,6-diamidino-2-phenylindole (DAPI, blue). Image representative of 3 experiments. (D) ApoE−/−;ΔdblGata+/+ mice (n = 10) showed enlarged areas of macrophages (green) relative to plaque area as well as areas of smooth muscle cells (red) compared with the ApoE−/−;ΔdblGATA1−/− mice (n = 9). (E) Left: Elevated CCL11 serum levels in ApoE−/− mice in response to 12 weeks on an HFD compared with a chow diet (n = 5 each). Right: Representative staining for CCL11 (green) and DAPI (nuclei in blue) in the aortic root of ApoE−/− mice after 12 weeks on an HFD. Dotted line indicates endothelium; solid line indicates internal elastic lamina. Scale bar, 50 µm. (F) Formation of eosinophil-platelet aggregates given as CD41+Siglec-F+ eosinophils of all eosinophils assessed by FACS (n = 5 with an HFD; n = 4 with a chow diet) in ApoE−/− mice after 12 weeks. (G) Platelets of ApoE−/−;ΔdblGATA1−/− mice (n = 5) adhered less often at the uninjured endothelium of the carotid artery under steady-state conditions compared with platelets of ApoE−/−;ΔdblGATA1+/+ mice (n = 6) as assessed by intravital microscopy. (H) Exposure of VWF on the endothelial surface of the intact carotid artery quantified by immunohistology in ApoE−/−;ΔdblGATA1−/− (n = 16) and ApoE−/−;ΔdblGATA1+/+ (n = 13) littermates after 13 weeks on an HFD. (I) Plasma levels of VWF in ApoE−/−;ΔdblGATA1−/− and ApoE−/−;ΔdblGATA1+/+ (n = 5 each) littermates after 13 weeks on an HFD. (J) Endothelial cells exposed VWF in response to supernatant of eosinophils activated with IL-5 and CCL11 compared with supernatant from resting eosinophils or medium with IL-5 and CCL11 (n = 4 each). Tumor necrosis factor α (TNF-α) was used as a positive control. For panels A-B and D-J, data are mean ± standard deviation (SD). *P < .05; **P < .01.
Eosinophils promote atherosclerotic plaque formation. (A) Atherosclerotic lesion formation (white dotted lines) assessed by lipid staining with Oil-Red O in the aortic root of eosinophil-deficient ApoE−/−;ΔdblGATA1−/− mice (n = 14: 7 female, 7 male) compared with littermate controls (ApoE−/−;ΔdblGATA+/+ (n = 13: 6 female, 7 male) after 13 weeks on an HFD. (B) Quantification of macroscopic lesion formation by Sudan III staining in the aortic arch of ApoE−/−;ΔdblGATA1−/− mice (n = 19: 9 female, 10 male) compared with ApoE−/−;ΔdblGATA+/+ littermate controls (n = 19: 9 female, 10 male). Scale bar, 200 µm. (C) Eosinophils were absent in plaques (white dotted line) of ApoE−/−;ΔdblGATA+/+ mice as assessed by staining for Siglec-F (green), nuclei are stained with 4′,6-diamidino-2-phenylindole (DAPI, blue). Image representative of 3 experiments. (D) ApoE−/−;ΔdblGata+/+ mice (n = 10) showed enlarged areas of macrophages (green) relative to plaque area as well as areas of smooth muscle cells (red) compared with the ApoE−/−;ΔdblGATA1−/− mice (n = 9). (E) Left: Elevated CCL11 serum levels in ApoE−/− mice in response to 12 weeks on an HFD compared with a chow diet (n = 5 each). Right: Representative staining for CCL11 (green) and DAPI (nuclei in blue) in the aortic root of ApoE−/− mice after 12 weeks on an HFD. Dotted line indicates endothelium; solid line indicates internal elastic lamina. Scale bar, 50 µm. (F) Formation of eosinophil-platelet aggregates given as CD41+Siglec-F+ eosinophils of all eosinophils assessed by FACS (n = 5 with an HFD; n = 4 with a chow diet) in ApoE−/− mice after 12 weeks. (G) Platelets of ApoE−/−;ΔdblGATA1−/− mice (n = 5) adhered less often at the uninjured endothelium of the carotid artery under steady-state conditions compared with platelets of ApoE−/−;ΔdblGATA1+/+ mice (n = 6) as assessed by intravital microscopy. (H) Exposure of VWF on the endothelial surface of the intact carotid artery quantified by immunohistology in ApoE−/−;ΔdblGATA1−/− (n = 16) and ApoE−/−;ΔdblGATA1+/+ (n = 13) littermates after 13 weeks on an HFD. (I) Plasma levels of VWF in ApoE−/−;ΔdblGATA1−/− and ApoE−/−;ΔdblGATA1+/+ (n = 5 each) littermates after 13 weeks on an HFD. (J) Endothelial cells exposed VWF in response to supernatant of eosinophils activated with IL-5 and CCL11 compared with supernatant from resting eosinophils or medium with IL-5 and CCL11 (n = 4 each). Tumor necrosis factor α (TNF-α) was used as a positive control. For panels A-B and D-J, data are mean ± standard deviation (SD). *P < .05; **P < .01.
Results
Eosinophils contribute to atherosclerotic lesion formation by activating endothelial cells
Eosinophils have been implicated in cardiovascular events, but it is unclear how they contribute to the inflammatory and thrombotic milieu. We first tested whether eosinophils influence the development of atherosclerotic lesions by using ApoE−/− mice crossed with eosinophil-deficient ΔdblGATA1−/− mice. These mice, as well as their wild-type littermates with functional eosinophils (ApoE−/−;ΔdblGATA1+/+), were fed a cholesterol-rich western diet for 13 weeks. In the aortic arch as well as at the level of the aortic root, eosinophil-deficient mice had markedly decreased lesion burden as assessed by lipid deposition compared with littermate controls; body weight, cholesterol levels, hemoglobin, and erythrocytes did not differ (Figure 1A-B; supplemental Figure 1A-D, available on the Blood Web site). However, as reported earlier for stable human atherosclerotic plaques, no eosinophils were found within murine lesions (Figure 1C).17 To study whether recruitment of other leukocyte populations was affected by the absence of eosinophils, we examined the inflammatory immune cell infiltration into plaques. ApoE−/−;ΔdblGATA1+/+ showed a higher percentage of neutrophils and macrophage area relative to atherosclerotic lesion area, and the percentage of smooth muscle cell area was also increased compared with that in eosinophil-deficient ApoE−/−;ΔdblGATA1−/− littermates (Figure 1D; supplemental Figure 1E-F). As expected, ApoE−/−;ΔdblGATA1−/− mice showed a systemic increase in monocyte and neutrophil counts in response to a high-fat diet (HFD) (supplemental Figure 1G). There was no difference in systemic monocyte counts in ApoE−/−;ΔdblGATA1−/− mice compared with wild-type littermates when both received HFDs. Neutrophils were slightly decreased in eosinophil-deficient mice (supplemental Figure 1H). These results suggest that eosinophils promote the inflammatory response in atherosclerosis and foster plaque formation that supports immune cell recruitment. In addition, their absence in the lesions indicates a systemic effect of eosinophils rather than a local effect within the plaque itself.
To test whether eosinophils are activated in the setting of atherosclerosis, we analyzed eosinophil-stimulating factors in ApoE−/− mice receiving an HFD or a chow diet. In response to an HFD, we found increased serum levels of CCL11 in ApoE−/− mice, which was also present within plaques and in the overlying endothelium (Figure 1E). Eosinophil blood counts were unchanged within the blood as well as in thrombi, and the major eosinophil mobilizing mediator IL-5 was only slightly but not significantly elevated (supplemental Figure 2A-C). However, eosinophils formed markedly more complexes with platelets, indicating that an interplay between these cell types might be involved in the promotion of atherosclerosis (Figure 1F). Because platelets are involved in atheroprogression,34,35 we hypothesized that eosinophils might amplify atherosclerosis by facilitating platelet adhesion. To test this in vivo, we assessed platelet recruitment to the carotid artery of ApoE−/−;ΔdblGATA1−/− under baseline conditions and measured platelet adhesion, which was markedly impaired in eosinophil-deficient mice (Figure 1G). However, platelets per se did not show any differences in terms of numbers, activation status, or platelet exposure of GPIb and VWF in ApoE−/−;ΔdblGATA1−/− mice compared with wild-type littermates. In addition, VWF exposure on platelets was not induced by activated eosinophils in vitro (supplemental Figure 2D-H). Another determinant of platelet adhesion is VWF exposure on the endothelium, and this was strongly decreased in eosinophil-deficient ApoE−/− mice in vivo, which also resulted in decreased plasma levels of VWF (Figure 1H-I). We confirmed in vitro that activated eosinophils induce exposure of VWF on endothelial cells, which was not the case for resting eosinophils (Figure 1J). In summary, these data suggest that eosinophils promote atherosclerosis involving VWF exposure on endothelial cells and increased atherogenic platelet adhesion to the vessel wall.
Eosinophils support arterial thrombosis by propagating platelet accumulation
Having shown that eosinophils support platelet adhesion and atheroprogression, we next asked whether they also support platelet accumulation after endothelial injury in the setting of atherosclerosis. To investigate this, we induced thrombosis in the carotid arteries of atherosclerotic eosinophil-deficient ApoE−/−;ΔdblGATA1−/− mice and littermate controls by applying FeCl3. The time between induction of thrombus formation and arterial occlusion was similar between eosinophil-deficient and wild-type littermates (supplemental Figure 3A). Strikingly, thrombi of eosinophil-deficient mice re-canalized earlier and were less stable. Consequently, the duration of complete arterial occlusion was significantly shorter in eosinophil-deficient mice compared with the littermate control group (Figure 2A; supplemental Video 1). Hence, eosinophils enhance thrombus stability during arterial thrombosis in the setting of atherosclerosis.
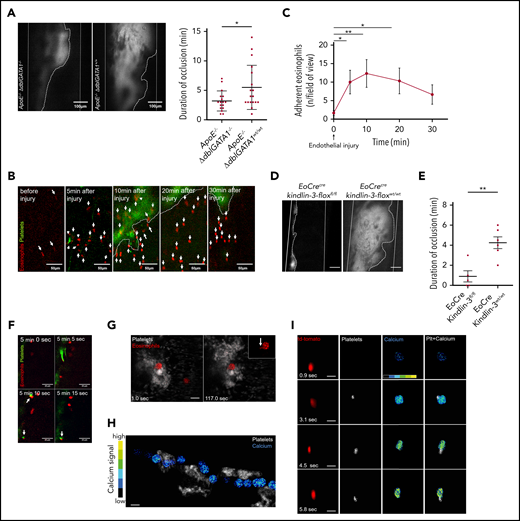
Eosinophil recruitment and platelet-induced activation in thrombosis. (A) Representative images of thrombus formation (marked by dotted line) in the carotid artery (marked by solid white lines) of an ApoE−/−;ΔdblGATA1−/− and an ApoE−/−;ΔdblGATA1+/+ mouse. In ApoE−/−;ΔdblGATA1−/− mice (n = 17), thrombus stability was significantly reduced compared with that of ApoE−/−;ΔdblGATA1+/+ littermate controls (n = 17), as shown by occlusion time. (B) Eosinophils (red, arrows) in EoCre;PC-G5-tdTfl/fl mice adhere immediately after initial endothelial injury at the lesion site of the carotid artery after FeCl3 application. Platelets labeled with fluorescent 2′,7′-dichlorofluorescein (green); thrombus shown by dotted line. (C) Quantification of eosinophil recruitment in vivo after FeCl3 application on the carotid artery (n = 3). (D) Representative images of thrombus formation 20 minutes after endothelial injury by FeCl3 in the carotid artery of EoCre;Kindlin-3fl/fl and EoCre;Kindlin-3wt/wt mice (thrombus area marked by dotted white line; carotid artery marked by solid white lines). Scale bar, 100 µm. (E) EoCre;Kindlin-3fl/fl mice (n = 5) had drastically reduced thrombus formation compared with that of wild-type littermates (n = 6). (F) During thrombus formation in the carotid artery, platelets (green) interacted with an eosinophil (red), which already adhered to the endothelium. Interacting cells are marked with arrows. (G) Eosinophils (red) in EoCre;PC-G5-tdTflox mice collect platelets (white) around themselves and form elongated processes (arrow in higher magnification, top right inset). Thrombosis induced by FeCl3 in the mesenteric microcirculation; images acquired by confocal microscopy. Scale bar, 20 µm. Images representative of 3 experiments. (H) Calcium signal (pseudocolored; blue/green, low signal; yellow/red, high signal) in an eosinophil interacting with several platelet aggregates (white) in the mesenteric microcirculation. Cropped images from intravital confocal microscopy in EoCre;PC-G5-tdTflox mice. Image representative of 3 experiments. (I) Calcium signal (pseudocolored) in an eosinophil (red) adhering to the endothelium before and during platelet contact (white). Image representative of 4 experiments. Cropped images from intravital epifluorescence microscopy in the carotid artery of EoCre;PC-G5-tdTflox mice after FeCl3 application. Scale bar, 20 µm. Representative of 4 experiments. For panels A-C and E, data are mean ± SD. *P < .05; **P < .01.
Eosinophil recruitment and platelet-induced activation in thrombosis. (A) Representative images of thrombus formation (marked by dotted line) in the carotid artery (marked by solid white lines) of an ApoE−/−;ΔdblGATA1−/− and an ApoE−/−;ΔdblGATA1+/+ mouse. In ApoE−/−;ΔdblGATA1−/− mice (n = 17), thrombus stability was significantly reduced compared with that of ApoE−/−;ΔdblGATA1+/+ littermate controls (n = 17), as shown by occlusion time. (B) Eosinophils (red, arrows) in EoCre;PC-G5-tdTfl/fl mice adhere immediately after initial endothelial injury at the lesion site of the carotid artery after FeCl3 application. Platelets labeled with fluorescent 2′,7′-dichlorofluorescein (green); thrombus shown by dotted line. (C) Quantification of eosinophil recruitment in vivo after FeCl3 application on the carotid artery (n = 3). (D) Representative images of thrombus formation 20 minutes after endothelial injury by FeCl3 in the carotid artery of EoCre;Kindlin-3fl/fl and EoCre;Kindlin-3wt/wt mice (thrombus area marked by dotted white line; carotid artery marked by solid white lines). Scale bar, 100 µm. (E) EoCre;Kindlin-3fl/fl mice (n = 5) had drastically reduced thrombus formation compared with that of wild-type littermates (n = 6). (F) During thrombus formation in the carotid artery, platelets (green) interacted with an eosinophil (red), which already adhered to the endothelium. Interacting cells are marked with arrows. (G) Eosinophils (red) in EoCre;PC-G5-tdTflox mice collect platelets (white) around themselves and form elongated processes (arrow in higher magnification, top right inset). Thrombosis induced by FeCl3 in the mesenteric microcirculation; images acquired by confocal microscopy. Scale bar, 20 µm. Images representative of 3 experiments. (H) Calcium signal (pseudocolored; blue/green, low signal; yellow/red, high signal) in an eosinophil interacting with several platelet aggregates (white) in the mesenteric microcirculation. Cropped images from intravital confocal microscopy in EoCre;PC-G5-tdTflox mice. Image representative of 3 experiments. (I) Calcium signal (pseudocolored) in an eosinophil (red) adhering to the endothelium before and during platelet contact (white). Image representative of 4 experiments. Cropped images from intravital epifluorescence microscopy in the carotid artery of EoCre;PC-G5-tdTflox mice after FeCl3 application. Scale bar, 20 µm. Representative of 4 experiments. For panels A-C and E, data are mean ± SD. *P < .05; **P < .01.
Next, we determined how eosinophils are recruited and activated in arterial thrombosis. To understand the dynamics of eosinophil recruitment in arterial thrombosis, we visualized thrombus development in EoCre;PC-G5-tdTfl/fl mice using intravital imaging. In this mouse model, eosinophils express the calcium indicator GCaMP5 emitting a green fluorescent protein signal upon binding of calcium in addition to a constitutively active tdTomato signal. Eosinophils adhered within a few minutes after FeCl3 application and were still present at the lesion site when imaging was stopped (Figure 2B-C). Eosinophil adhesion has been reported to involve integrins, so we tested whether eosinophils were just passively trapped in the thrombus or actively recruited.36,37 Eosinophils were markedly enriched in arterial thrombi compared with blood, arguing against a passive process (supplemental Figure 3B). In addition, when we examined thrombus development in EoCre;Kindlin-3fl/fl mice, in which eosinophils lack the pan-integrin regulatory protein Kindlin-3, we found significantly shorter durations of occlusion, and formation of occlusive thrombi was severely impaired. This indicates that integrins are essential for local recruitment and activation of eosinophils in arterial thrombosis (Figure 2D-E; supplemental Video 2). Because platelets were found to activate eosinophils in the setting of allergic reactions, we evaluated whether platelets directly interact with and activate eosinophils in vivo.38,39 Indeed, we found frequent interactions between platelets and eosinophils during thrombus formation. Eosinophils served as a nidus for thrombus formation by bundling platelets around them. These interactions ranged from short-term to permanent contacts of eosinophils and platelets, resulting in a shape change for eosinophils which exposed elongated processes (Figure 2F-G; supplemental Video 3). Platelet-eosinophil interactions also had functional effects that resulted in markedly increased calcium activity when we tracked the calcium signal in eosinophils in vivo in EoCre;PC-G5-tdTfl/fl mice: eosinophils circulating in the bloodstream showed calcium bursts when they encountered platelet aggregates attached to the injury site (Figure 2H; supplemental Figure 3C; supplemental Video 4). During firm adhesion to the vessel wall, eosinophils showed a short calcium transient which rapidly returned to a low calcium signal. However, when adhering eosinophils encountered platelets, they had a marked increase in calcium signal. Intracellular calcium was persistently elevated compared with eosinophils which stayed at the vessel wall without contacting platelets (Figure 2I; supplemental Figure 3D-F). In turn, activated eosinophils were able to induce platelet aggregation in vitro (supplemental Figure 3G). These findings indicate that eosinophils are recruited after endothelial injury in an integrin-dependent manner to the site of thrombus formation, where a crosstalk with platelets results in mutual activation.
Direct interaction with platelets induces eosinophil activation and EET release
We next defined the communication signals used by platelets to activate eosinophils in vitro. To assess eosinophil activation, we measured IL-5 secretion, which enhances their recruitment and activation in an autocrine manner.40 Thrombin-activated platelets, but not the cell-free supernatant of activated platelets, induced IL-5 secretion by eosinophils (Figure 3A). Importantly, resting platelets had only a minor effect on eosinophil IL-5 release.
![Platelets trigger EET formation. (A) Eosinophils release IL-5 after co-incubation with activated platelets (plt) (n = 8). Resting platelets (n = 8), supernatant (conditioned medium [CM]) of activated platelets (activ. plt) (n = 6), and thrombin (n = 4) had significantly smaller effects on IL-5-secretion of eosinophils. (B) Isolated eosinophils formed EETs containing MBP (arrows) after activation by platelets. EETs were also found in human arterial thrombi and in murine arterial thrombi. Images representative of 5 experiments. (C) Quantification of EETs and NETs in thrombi induced by FeCl3 in ApoE−/− mice (n = 5) after 13 weeks of HFD given as percent of total extracellular traps. (D) Eosinophils formed EETs after interaction with activated platelets (n = 8), whereas resting platelets (n = 7), CM (n = 6), and thrombin control (n = 8) lead to significantly less EET formation. (E) Treatment of eosinophils and platelets with a P-selectin blocking antibody before activation inhibited EET formation (n = 7) compared with isotype control (n = 7). (F) Representative images of EET formation. Arrows indicate EETs. (G) CCL5-blocking antibodies failed to reduce EET formation (n = 6) compared with isotype control (n = 6). (H) In vitro experiments with isolated human eosinophils showed that EET formation declined when eosinophils were pretreated with Siglec-8 binding antibodies (n = 5) compared with EET formation in cells treated with isotype control antibodies (n = 7) before activation by platelets. (I) The images show representatively that Siglec-8 binding antibodies prevent EET formation (arrows). For panels A, C-E, and G-H, data are mean ± SD. *P < .05; **P < .01; ****P < .0001. ns, not significant.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/134/21/10.1182_blood.2019000518/7/m_bloodbld2019000518f3.png?Expires=1769092524&Signature=b3oAmequahQgI~dyqJSR-vpVZ4tnNVOf5evLOvsay~4Y-VDyct28xOynRcPSUcV~KmA0IEIBeprVCC0TEX4sEXXPHqEIQE0GwcsY5r5Pc5Fe2MaFWOoxMMppVZ7kWyBuP9QEpJj1za-B3VhEhIPGnFIPAJzPGlf-dt0aMrNyJlZue-3PW91pP~dR2F4f21gJY-vJtxCRnH~a01DfNMgUH9zXAaE7DLPJPpGJU7TtCFpUezO-XLNdL4ZBP1a6lwm4q8O3XpBoo3cyKlvN3fT53oWFQo6EO1t0qfVmCPF3S7dAoZTAphvsVmNLe2rmi3teda4cEgImDPz7EniNpgp5~g__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Platelets trigger EET formation. (A) Eosinophils release IL-5 after co-incubation with activated platelets (plt) (n = 8). Resting platelets (n = 8), supernatant (conditioned medium [CM]) of activated platelets (activ. plt) (n = 6), and thrombin (n = 4) had significantly smaller effects on IL-5-secretion of eosinophils. (B) Isolated eosinophils formed EETs containing MBP (arrows) after activation by platelets. EETs were also found in human arterial thrombi and in murine arterial thrombi. Images representative of 5 experiments. (C) Quantification of EETs and NETs in thrombi induced by FeCl3 in ApoE−/− mice (n = 5) after 13 weeks of HFD given as percent of total extracellular traps. (D) Eosinophils formed EETs after interaction with activated platelets (n = 8), whereas resting platelets (n = 7), CM (n = 6), and thrombin control (n = 8) lead to significantly less EET formation. (E) Treatment of eosinophils and platelets with a P-selectin blocking antibody before activation inhibited EET formation (n = 7) compared with isotype control (n = 7). (F) Representative images of EET formation. Arrows indicate EETs. (G) CCL5-blocking antibodies failed to reduce EET formation (n = 6) compared with isotype control (n = 6). (H) In vitro experiments with isolated human eosinophils showed that EET formation declined when eosinophils were pretreated with Siglec-8 binding antibodies (n = 5) compared with EET formation in cells treated with isotype control antibodies (n = 7) before activation by platelets. (I) The images show representatively that Siglec-8 binding antibodies prevent EET formation (arrows). For panels A, C-E, and G-H, data are mean ± SD. *P < .05; **P < .01; ****P < .0001. ns, not significant.
Platelets trigger EET formation. (A) Eosinophils release IL-5 after co-incubation with activated platelets (plt) (n = 8). Resting platelets (n = 8), supernatant (conditioned medium [CM]) of activated platelets (activ. plt) (n = 6), and thrombin (n = 4) had significantly smaller effects on IL-5-secretion of eosinophils. (B) Isolated eosinophils formed EETs containing MBP (arrows) after activation by platelets. EETs were also found in human arterial thrombi and in murine arterial thrombi. Images representative of 5 experiments. (C) Quantification of EETs and NETs in thrombi induced by FeCl3 in ApoE−/− mice (n = 5) after 13 weeks of HFD given as percent of total extracellular traps. (D) Eosinophils formed EETs after interaction with activated platelets (n = 8), whereas resting platelets (n = 7), CM (n = 6), and thrombin control (n = 8) lead to significantly less EET formation. (E) Treatment of eosinophils and platelets with a P-selectin blocking antibody before activation inhibited EET formation (n = 7) compared with isotype control (n = 7). (F) Representative images of EET formation. Arrows indicate EETs. (G) CCL5-blocking antibodies failed to reduce EET formation (n = 6) compared with isotype control (n = 6). (H) In vitro experiments with isolated human eosinophils showed that EET formation declined when eosinophils were pretreated with Siglec-8 binding antibodies (n = 5) compared with EET formation in cells treated with isotype control antibodies (n = 7) before activation by platelets. (I) The images show representatively that Siglec-8 binding antibodies prevent EET formation (arrows). For panels A, C-E, and G-H, data are mean ± SD. *P < .05; **P < .01; ****P < .0001. ns, not significant.
Besides releasing IL-5, activated eosinophils form extracellular DNA structures (EETs), the eosinophil counterparts to NETs.41,42 Recently, NETs were found to contribute to thrombus formation by activating platelets.10,12,43 The effect of NETs on activation of the intrinsic pathway of coagulation is controversial; in vivo studies suggest a potentially indirect effect, but in vitro studies found no direct effect.44 When we evaluated murine arterial thrombi, we found EETs that could be differentiated from NETs by extracellular DNA covered with MBP, which indicated their eosinophilic origin. In addition, with 27% of extracellular traps originating from eosinophils and 67% originating from neutrophils, EETs make a substantial contribution. The presence of EETs was also confirmed in human coronary artery thrombi (Figure 3B-C). To determine the trigger for EET formation, we co-incubated isolated eosinophils with platelets and measured EET formation. Indeed, eosinophils formed EETs after contact with activated platelets, but not after incubation with their supernatant or with resting platelets (Figure 3D). This suggests that direct platelet-eosinophil interactions are essential for eosinophil activation by platelets. To test this, we disrupted this interaction by using an anti-P-selectin antibody, which resulted in reduced EET formation (Figure 3E-F).45 We also assessed the effect of blocking CCL5, an eosinophil-activating chemokine shed by activated platelets, but this had no effect on EET formation (Figure 3G).39 Recently, antibodies to the eosinophil inhibitory receptor Siglec-8 and the murine homolog Siglec-F were found to be potent and selective inhibitors of eosinophils. This mechanism is considered for therapeutic interventions in eosinophil-mediated diseases.46,47 Indeed, we found that treatment with an anti-Siglec-8 antibody impaired EET formation (Figure 3H-I). Therefore, eosinophils are activated through direct contacts with platelets, which trigger EET formation.
EET formation stabilizes arterial thrombi
Having shown that EETs are induced by platelets, we next determined the importance of EETs for arterial thrombosis. For this, we wondered whether Siglec-F–mediated direct inhibition of eosinophils might be a novel therapeutic approach. Indeed, mice treated with Siglec-F antibodies 20 minutes before thrombosis showed significantly reduced thrombus stability compared with the control group (Figure 4A-B; supplemental Video 5). Because Siglec-F antibodies have a long-term depleting effect, we assessed whether this short-term effect was primarily caused by eosinophil depletion or whether it involved eosinophil inactivation. We did not observe any significant differences in eosinophil numbers systemically in the blood or locally in the thrombi between Siglec-F antibody and its isotype (Figure 4C-D). Platelet counts in peripheral blood were comparable in these 2 groups (supplemental Figure 4A-B). Because these observations indicate that anti-Siglec-F antibodies reduce thrombus stability independent from their depleting effect and because EET formation is decreased after anti-Siglec-F antibody treatment in vitro, we determined EET formation in thrombi. In line with our in vitro findings, we detected significantly reduced EET formation in mice treated with the anti-Siglec-F antibody (Figure 4E). Therefore, targeting EET formation might be a new approach for preventing arterial thrombosis.
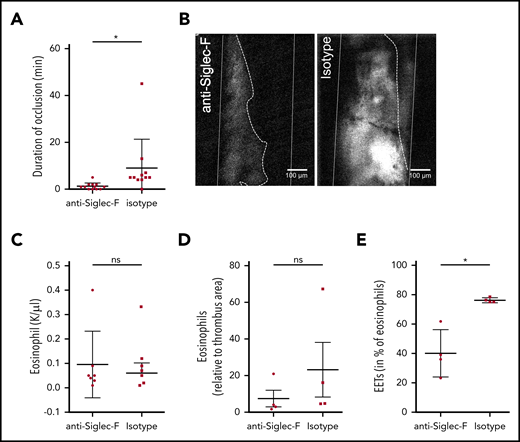
Targeting EET formation reduces thrombus formation. (A) C57BL6 mice, treated with anti-Siglec-F antibodies (n = 12) did not develop long-lasting, occlusive thrombi after FeCl3 application to the carotid artery. Treatment with an isotype-control (n = 11) did not impair thrombus development. (B) Representative image of thrombus formation 20 minutes after initial endothelial damage of a mouse treated with Siglec-F antibody and control mouse (thrombus area marked with white dotted line; artery walls marked with white lines; recanalization marked with dotted line). (C-D) Mice treated with anti-Siglec-F and isotype control show no differences in numbers of eosinophils in blood or in thrombi. (E) The number of eosinophils forming EETs in thrombi of mice treated with Siglec-F antibody (n = 4) decreased significantly compared with isotype control (n = 4). For panels A and C-E, data are mean ± SD. *P < .05.
Targeting EET formation reduces thrombus formation. (A) C57BL6 mice, treated with anti-Siglec-F antibodies (n = 12) did not develop long-lasting, occlusive thrombi after FeCl3 application to the carotid artery. Treatment with an isotype-control (n = 11) did not impair thrombus development. (B) Representative image of thrombus formation 20 minutes after initial endothelial damage of a mouse treated with Siglec-F antibody and control mouse (thrombus area marked with white dotted line; artery walls marked with white lines; recanalization marked with dotted line). (C-D) Mice treated with anti-Siglec-F and isotype control show no differences in numbers of eosinophils in blood or in thrombi. (E) The number of eosinophils forming EETs in thrombi of mice treated with Siglec-F antibody (n = 4) decreased significantly compared with isotype control (n = 4). For panels A and C-E, data are mean ± SD. *P < .05.
Eosinophils release MBP and enhance thrombus formation
We then sought to determine how EETs contribute to the development of arterial thrombosis. Previous studies showed that the eosinophil cytotoxic protein MBP and EPX can activate platelets in vitro.48 To determine whether platelets can activate eosinophils to secrete these granule proteins, we examined platelet-induced EETs by confocal microscopy and found colocalization of platelets and MBP on EETs (Figure 5A). Next, we determined the in vivo relevance of MBP and EPX in arterial thrombosis using MBP- (MBP−/−) and EPX-deficient mice (EPX−/−) in comparison with wild-type littermates. Strikingly, mice lacking MBP had substantially decreased thrombus stability with decreased durations of complete occlusion (Figure 5B-C; supplemental Video 6). Thrombi in MBP−/− mice failed to fully occlude the vessel, but systemic eosinophil and platelet counts were not affected (supplemental Figure 5A-C). In contrast, the other cytotoxic protein EPX had no effect in this model of arterial thrombosis (Figure 5D). Previously, in vitro studies showed MBP binding to thrombomodulin and an inhibitory effect on APC formation, but APC levels in MBP−/− mice were not altered compared with those in wild-type littermates (Figure 5E).49 To further determine the thrombus-stabilizing effect of MBP, we activated eosinophils in whole blood of MBP−/− mice with IL-5 and CCL11 followed by ADP-induced platelet aggregation. Interestingly, platelets in whole blood of MBP−/− mice showed significantly less aggregation, indicating that MBP exposed on EETs enhances platelet activation and promotes thrombus development (Figure 5F).
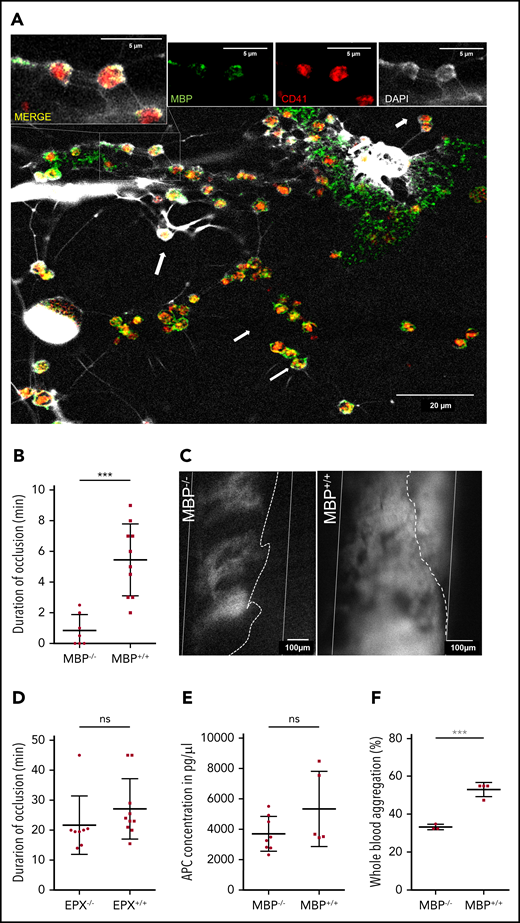
Major basic protein on EETs activates platelets. (A) Eosinophils activated in vitro by platelets (red) formed EETs containing MBP (green) and DNA (white, DAPI). On EETs, MBP and platelets colocalize (arrows) as shown in higher magnification in the insets at the top of the panel. Images from confocal microscopy representative of 3 experiments. (B) Thrombus formation in MBP−/− mice (n = 8) is significantly impaired compared with thrombus formation in MBP+/+ littermates (n = 10). (C) Representative images of platelet aggregation (gray area, dotted line) in the carotid artery (white lines) of MBP−/− and normal thrombus development in MBP+/+ mice 20 minutes after endothelial injury. (Recanalization is marked by dotted line). (D) EPX−/− mice (n = 8) as well as EPX+/+ (n = 10) mice showed occlusive thrombus formation. (E) APC concentrations in plasma of MBP+/+ (n = 5) and MBP−/− (n = 8) did not show any differences after arterial thrombosis. (F) Platelet aggregation induced by ADP in IL-5 and eotaxin-treated whole blood of female MBP−/− mice (n = 4) was significantly decreased compared with that in wild-type mice (n = 3). For panels B and D-F, data are mean ± SD. ***P < .001.
Major basic protein on EETs activates platelets. (A) Eosinophils activated in vitro by platelets (red) formed EETs containing MBP (green) and DNA (white, DAPI). On EETs, MBP and platelets colocalize (arrows) as shown in higher magnification in the insets at the top of the panel. Images from confocal microscopy representative of 3 experiments. (B) Thrombus formation in MBP−/− mice (n = 8) is significantly impaired compared with thrombus formation in MBP+/+ littermates (n = 10). (C) Representative images of platelet aggregation (gray area, dotted line) in the carotid artery (white lines) of MBP−/− and normal thrombus development in MBP+/+ mice 20 minutes after endothelial injury. (Recanalization is marked by dotted line). (D) EPX−/− mice (n = 8) as well as EPX+/+ (n = 10) mice showed occlusive thrombus formation. (E) APC concentrations in plasma of MBP+/+ (n = 5) and MBP−/− (n = 8) did not show any differences after arterial thrombosis. (F) Platelet aggregation induced by ADP in IL-5 and eotaxin-treated whole blood of female MBP−/− mice (n = 4) was significantly decreased compared with that in wild-type mice (n = 3). For panels B and D-F, data are mean ± SD. ***P < .001.
Enrichment of eosinophils in female patients with ST
To assess whether specific patient populations have different numbers of eosinophils, we systematically examined human coronary artery thrombi for the presence of eosinophils. For that, we compared the numbers of eosinophils in thrombi according to the setting of thrombosis (acute myocardial infarction [AMI] of native vessels or ST), sex, cardiovascular risk factors, and clinical presentation. Indeed, eosinophils were detected by Luna staining in all thrombi (Figure 6A). There was no difference in eosinophil numbers between the AMI and ST groups (data not shown). However, logistic regression analysis of thrombi revealed significantly elevated numbers of eosinophils in thrombi from female patients who had an ST (Figure 6B-C). There was no association of eosinophils with cardiovascular risk factors (Figure 6B). Furthermore, clinical presentation with an ST-elevated myocardial infarction or a non-ST elevated myocardial infarction did not influence the number of eosinophils in human coronary artery thrombi (Figure 6B). In the subgroup of female patients with ST, no influence of cardiovascular risk factors on eosinophil numbers was identified (Figure 6D). Therefore, the contribution of eosinophils to thrombosis might depend on sex, and female patients with ST showed the highest numbers of eosinophils in coronary artery thrombi.
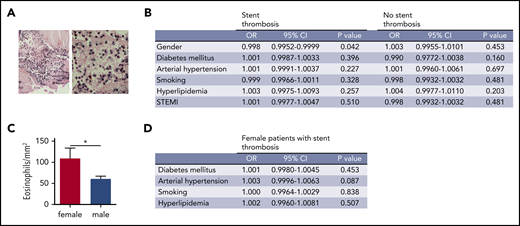
Eosinophil numbers in thrombi from patients with myocardial infarction. (A) Representative image of a Luna staining of a human coronary artery thrombus. Left: overview image. Original magnification ×20. Right: higher magnification (original magnification ×40) showing eosinophils (arrowheads) with polylobulated nuclei (dark blue) and eosinophil granules (bright red). Scale bar, 75 µm. (B) Logistic regression analysis of eosinophil numbers in coronary artery thrombi divided by type of thrombosis (ST vs native vessel thrombosis) according to gender, diabetes mellitus, arterial hypertension, smoking status, hyperlipidemia, or presentation as ST-elevation myocardial infarction. (C) Eosinophils/mm2 in human coronary artery thrombi from male (n = 168) and female patients (n = 48) with ST. (D) Logistic regression analysis of eosinophil numbers in coronary artery thrombi from female patients with ST according to diabetes mellitus, arterial hypertension, smoking status, and hyperlipidemia. CI, confidence interval; OR, odds ratio.
Eosinophil numbers in thrombi from patients with myocardial infarction. (A) Representative image of a Luna staining of a human coronary artery thrombus. Left: overview image. Original magnification ×20. Right: higher magnification (original magnification ×40) showing eosinophils (arrowheads) with polylobulated nuclei (dark blue) and eosinophil granules (bright red). Scale bar, 75 µm. (B) Logistic regression analysis of eosinophil numbers in coronary artery thrombi divided by type of thrombosis (ST vs native vessel thrombosis) according to gender, diabetes mellitus, arterial hypertension, smoking status, hyperlipidemia, or presentation as ST-elevation myocardial infarction. (C) Eosinophils/mm2 in human coronary artery thrombi from male (n = 168) and female patients (n = 48) with ST. (D) Logistic regression analysis of eosinophil numbers in coronary artery thrombi from female patients with ST according to diabetes mellitus, arterial hypertension, smoking status, and hyperlipidemia. CI, confidence interval; OR, odds ratio.
Discussion
Arterial thrombosis is the most severe consequence of atherosclerosis, and both diseases are triggered and supported by sterile inflammation. Clinical observations suggest that eosinophils are involved in atherosclerotic plaque formation and arterial thrombosis, but the underlying mechanisms are incompletely understood.19,21,23,32,50 Here, we show that eosinophils are activated in the setting of atherosclerosis and they foster plaque formation. This is associated with augmented VWF exposure on the endothelium as well as enhanced platelet adhesion and immune cell recruitment into the vessel wall. Once arterial thrombosis is triggered, eosinophils are rapidly recruited to the lesion site in an integrin-dependent manner and intensively interact with platelets resulting in propagation and stabilization of developing thrombi. This leads to eosinophil activation as we demonstrate by real-time calcium imaging in vivo. Platelets stimulate eosinophils to form EETs which in turn induce platelet activation by the granule protein MBP covering the surface of these DNA structures. Targeting EET formation in vivo proved to be a valuable approach for preventing arterial thrombosis. From a clinical perspective, this might be particularly relevant for the subgroups of female patients who have coronary ST as identified in our prospective registry that enrolled patients with myocardial infarction. Taken together, our data demonstrate that eosinophils can contribute to cardiovascular events by participating in atherosclerosis as well as in arterial thrombosis through an intense interaction with platelets.9
Eosinophils are considered to be important mainly in allergic reactions and host defense against parasites. Although they have also been associated with atherosclerosis, they were not detected within stable atherosclerotic plaques in humans, which is in line with our findings in mice.17 However, the chemokine RANTES, known to be chemotactic to eosinophils, is deposited on plaques by circulating platelets, which exacerbate atherosclerosis through chemokine shedding.34,39,51 In addition, CCL11, a known specific chemoattractant and activator of eosinophils, is highly overexpressed in atherosclerotic plaques, which is confirmed in our study in mice.15 We demonstrate in the well-established eosinophil-deficient ΔdblGATA1−/− mouse strain that atherosclerosis is markedly diminished in the absence of eosinophils, but we did not investigate another eosinophil-deficient strain such as PHIL mice.52 How do eosinophils promote plaque formation and local inflammation if they are not recruited into the vessel wall? This effect could be a result of shedding of eosinophil granule proteins, namely MBP and ECP, whose blood concentration is elevated in atherosclerosis.14 In humans, elevated plasma levels of ECP are a predictor of atherothrombotic events and development of advanced atherosclerotic lesions.23 These data suggest that RANTES and CCL11 might recruit and activate eosinophils in atherosclerosis, and we show here increased serum levels of CCL11 in response to HFD in ApoE−/− mice. Eosinophilic degranulation products are cytotoxic and have a high potential to damage and activate endothelial cells.53 In line with this, we provide evidence that eosinophils trigger exposure of VWF on endothelial cells. In turn, eosinophil deficiency in atherosclerosis is associated with reduced endothelial VWF exposure and atherogenic platelet adhesion in vivo. However, this might be an indirect effect that results from the altered inflammatory milieu of reduced atherosclerosis or a decrease in neutrophils. The atherogenic effect of platelets depends on adhesion molecules such as GPIIb and especially GPIb α interacting with VWF.54-56 Interference with these platelet adhesion molecules as well as deletion of VWF results in attenuated atherosclerotic plaque formation.57-60 At the endothelium, platelets exert proinflammatory effects that trigger the recruitment of leukocytes through mediators such as RANTES.61 In summary, eosinophils promote atherosclerotic lesion formation, which is associated with increased platelet adhesion and leukocyte recruitment.
In addition, eosinophils and EETs are present in ruptured human atherosclerotic plaques as well as arterial thrombi and therefore might contribute to these processes in patients.16 Eosinophilia in autoimmune diseases, parasitic infections, and hypereosinophilic syndrome is associated with increased risk of arterial thrombosis, which is a platelet-driven process.62-67 In line with this, we found a stabilizing effect of eosinophils on thrombus formation in the setting of atherosclerosis. There, eosinophils accumulate in high numbers on the injured vessel wall. We found that integrin-mediated recruitment is crucial for the thrombosis-promoting effect of eosinophils, because Kindlin-3–deficient eosinophils no longer enhanced thrombus formation. Moreover, platelets are important activators of eosinophils, and we provide evidence that direct interactions are mandatory for platelet-induced IL-5 secretion (an interleukin found in elevated levels in patients with unstable angina and AMI50 ) and EET formation. Using intravital microscopy combined with real-time calcium imaging, we show for the first time that platelets provide an activating signal to eosinophils in vivo. Under shear flow conditions in vitro, platelets induce eosinophil rolling through PSGL-1/P-selectin interactions, but firm adhesion of eosinophils requires β2-integrin activation.68 The former pathway is also involved in platelet-induced eosinophil activation in vitro. The activation of eosinophils results in the formation of EETs, which constitute a substantial part of extracellular traps in murine thrombi and are also present in human thrombi. As we show here, these can be targeted therapeutically in vivo by using an antibody directed against Siglec-8, which is also involved in β2-integrin–dependent functions of eosinophils.47,69 EETs are decorated with MBP, which is activated in the extracellular space by forming aggregates, which are also seen on EETs70 and have been shown to bind thrombomodulin and block thrombin inactivation. Furthermore, through this pathway, APC formation was successfully inhibited in vitro.49,71 Surprisingly, MBP−/− mice had no elevated APC levels in thrombosis compared with littermate controls. However, the impaired thrombus formation in MBP−/− mice and subsequently less thrombin and APC formation could obscure this local mechanism on a systemic level. In addition, MBP was found to activate platelets in vitro by inducing the release of serotonin as well as α and dense granules, which increase platelet activation in an autocrine manner.48 We confirmed the effect of MBP in vivo on thrombus formation and in vitro on platelet aggregation, but could not find an effect of EPX in vivo. Eosinophils also contribute to thrombus formation by delivering activated TF, but the in vivo relevance of this procoagulant effect has only been shown in a model of venous thrombosis.22,23 Hence, besides their procoagulant effects, eosinophils promote arterial thrombosis by EET formation and MBP release resulting in platelet activation.
In conclusion, we demonstrate a self-reinforcing interplay of platelets and eosinophils in atherosclerosis and thrombosis in vivo and identify EET formation as a critical factor for thrombus stability. The clinical importance of these findings is supported by the accumulation of high numbers of eosinophils and EETs in coronary artery thrombi, in which female patients with ST showed the highest eosinophil counts. Therefore, this subgroup of patients might benefit most from an eosinophil-inhibiting strategy.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
The online version of this article contains a data supplement.
Acknowledgments
The authors thank Nicole Blount, Beate Jantz, and Sebastian Helmer for excellent technical assistance, and James Lee, who sadly died, for his valuable input to this project and for providing mice.
This study was supported by the Deutsche Forschungsgemeinschaft through the collaborative research center 1123 project A07 (K.S.), collaborative research center 914 project B02 (K.S.) and project A01 (M.M.); Deutsche Gesellschaft für Kardiologie (J.N.); LMUexcellent (K.S.); the European Union Seventh Framework Programme FP7/2007-2013 under grant No. HEALTH-F2-2010-260309 (PRESTIGE) (J.N.); and the Deutsche Zentrum für Herz-Kreislauf-Forschung (postdoctoral start-up grant [K.S.] and Clinician Scientist programme [L.N.]).
Authorship
Contribution: C.M. performed in vivo thrombosis experiments and in vitro experiments with eosinophils, platelets, and endothelial cells and planned the experiments; J.N. analyzed patient data and planned the experiments; D.S. and K.P. analyzed atherosclerotic plaques; K.R.Z. provided MBP, EPX, and EoCre mice; L.N. performed intravital confocal microscopy; T.J.S. supported approval of animal experiments; B.K., L.T.W., and J.P. performed intravital microscopy; D.K. performed logistic regression analysis; M.J. analyzed coronary artery thrombi; W.D., T.A., F.-J.N., A.H.G., and J.M.t.B. collected and provided human arterial thrombi; M.M. provided Kindlin3fl/fl mice; M.L. and B.K. performed in vitro experiments; S.S. and N.B. performed histology; S.S. and J.P. performed platelet studies; and K.S. designed the experiments and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Konstantin Stark, Medizinische Klinik und Poliklinik I, Klinikum der Universität München, Marchioninistr 15, 81377 Munich, Germany; e-mail: konstantin.stark@med.uni-muenchen.de.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal