

Abstract
Hemophilia A (HA) and hemophilia B (HB) are the most common severe bleeding disorders. Replacement therapy, providing the missing coagulation factor, has been the mainstay of treatment both prophylactically and to treat bleeding. Despite widespread availability of safe and effective replacement therapy, patients with HA and HB continue to experience a tremendous burden of treatment, breakthrough bleeding, and progressive joint disease, as well as high rates of inhibitor development. These remaining challenges are now being addressed by incredible advances in bioengineering. Recombinant bioengineering has led to replacement therapies with easier modes of administration, decreased immunogenicity, increased efficacy, and extended half-lives. Emicizumab, a bispecific antibody that acts as a substitutive therapy for HA, has been approved for patients with and without inhibitors. Novel compounds are in development to exploit the natural balance of hemostasis by targeting the natural anticoagulants protein C, protein S, tissue factor pathway inhibitor, and antithrombin. The substitution and rebalancing therapies provide an opportunity for steady-state hemostatic control without exposure to immunogenic clotting factor proteins. As such, they may have broader applications outside those being investigated in the clinical trial programs.
Introduction
Factor VIII (FVIII) deficiency (hemophilia A [HA]) and FIX deficiency (hemophilia B [HB]) are the most common severe bleeding disorders.1 Severe disease, <1% residual factor activity, leads to bleeding manifestations often without recognized trauma and are primarily into joints (hemarthrosis) but may include bleeding into muscles, soft tissues, and life-threatening locations (eg, central nervous system).2 Recurrent hemarthroses lead to hypertrophic synovitis, progressive cartilage degradation, and the long-term sequelae of hemophilic arthropathy with chronic pain and joint function impairment. Widespread availability of safe and efficacious factor products that can prevent bleeds and preserve healthy joints, as first pioneered in Sweden,3 allowed patients to be shifted from episodic treatment to prophylaxis
The need for continued innovation
The implementation of prophylaxis, beginning as early as the first year of life, is associated with significant treatment burden and cost. Replacement therapy has also been compromised by the development of alloantibodies to FVIII and FIX. These occur in ∼25% to 40% of patients with severe HA within the first 50 exposure days to FVIII, neutralizing its function.4 Such patients require treatment with bypassing agents (BPAs)5 and immune tolerance induction (ITI) to eradicate inhibitors, but patients continue to exhibit increased morbidity and mortality.6 In noninhibitor patients, prophylaxis can prevent joint bleeding and joint disease. However, studies have shown that bleeding events are not eliminated for all patients, and joint disease still appears in young adults.7-9 Given an expected normal lifespan, this cohort may still experience progressive joint disease over their lifetime.
Bioengineering innovations are addressing these challenges. The past 5 years have seen the approval of new recombinant products with extended half-lives, allowing less frequent infusions and/or better hemostatic control.10 Some new recombinant FVIII (rFVIII) products may exhibit reduced immunogenicity and more rapid responses to ITI. These products have been the subject of many reviews.11-13 We are now on the cusp of a paradigm shift in prophylaxis for hemophilia through the development of the first substitution therapy not based on the FVIII molecule, as well as innovative therapies that seek to rebalance the hemostatic defect by targeting the natural anticoagulant pathways that regulate hemostasis. Investigational gene therapy also shows promise for a definitive clinical cure and is covered in an accompanying review.
Novel FVIII products
The plasma half-life of the standard therapies for hemophilia requires frequent administration within prophylaxis regimens. These are typically given 3 times per week to every other day for FVIII and 2 or 3 times per week for FIX. Additionally, all current rFVIII products are limited by their IV mode of administration. Subcutaneous dosing may reduce the treatment burden, especially in those with difficult access, such as infants and toddlers. N8-GP is a glycoPEGylated extended half-life (EHL) rFVIII currently in late clinical studies. Preclinical studies in mice and monkeys, coupled with a human pharmacokinetic (PK) model, predicted FVIII trough levels of 2.5% to 10% with daily subcutaneous dosing.14
The EHL factor products have shown benefit by reducing the burden of prophylaxis through reduced frequency of administration and improving adherence, as well as by the ability to achieve and maintain higher trough levels for more effective bleeding prevention.10 rFVIII fused to the Fc component of immunoglobulin (rFVIIIFc) and PEGylation of rFVIII have shown a modest impact on their half-life (up to 1.5-fold), likely secondary to interaction with endogenous von Willebrand factor (VWF) and clearance as part of the VWF–FVIII complex.15 BIVV001 is a novel fusion protein that consists of a single-chain rFVIIIFc fused to the FVIII-binding D′D3 domains of VWF, as well as 2 XTEN linkers, unstructured polypeptides that reduce the rate of clearance and degradation.16 In animal studies, BIVV001 has been shown not to bind endogenous VWF and achieves a fourfold extension of half-life compared with standard products. In addition, the presence of the D′D3 domains and 2 XTEN linkers increases subcutaneous bioavailability to 20%, up from <1% with standard FVIII. Despite this, only the IV formulation is moving forward to clinical studies. Preliminary data from an ongoing phase 1/2a study in adults with severe HA showed a half-life of 37 hours compared with 13 hours with standard FVIII, with an average residual FVIII level of 13% at 5 days and 5.6% at 7 days post-IV infusion and no inhibitors.17 Such PKs would allow for once weekly prophylaxis. A novel strategy for ultrastable FVIII complexing with VWF is being investigated using nanobody technology. Nanobodies are based on naturally occurring antibodies identified in camels and llamas that are fully functional: they consist of heavy chains, lack light chains, and possess a single variable domain. These cloned single-variable domains have full antigen-binding capacity, are stable, and can be used to engineer biologics, nanobodies that include their unique affinity characteristics.18 Researchers engineered 2 copies of a noninhibitory nanobody that recognizes the VWF D′D3 domain with slow dissociation kinetics and replaced the FVIII B domain, such that it would be liberated upon cleavage by thrombin19 (Figure 1). This variant, FVIII-KB013bv, was fully active in 1-stage and chromogenic FVIII activity assays, had 25-fold higher affinity for VWF compared with B domain–deleted FVIII (BDD-FVIII), and, in a mouse model, exhibited a twofold prolongation of plasma half-life and increased efficacy in a tail-clip bleeding assay. Furthermore, the immune response to FVIII-KB013bv was significantly reduced compared with BDD-FVIII, with the hypothesis being that, given the high affinity of FVIII-KB013bv for VWF, it does not dissociate upon interaction of the complex with cellular antigen-presenting cells. This reduces FVIII uptake and the number of FVIII peptides available for presentation to T cells, thus reducing anti-FVIII antibody formation. Other novel recombinant products may also have reduced immunogenicity and improve attempts at eradication of inhibitors.20,21 Two new rFVIII products (human-cl rhFVIII and rFVIIIFC) are produced in human cell lines. Human-cl rhVIII has been shown to contain only human posttranslational modifications and a high degree of sulfation at Tyr740, eliminating potentially immunogenic nonhuman glycan epitopes and enhancing affinity for VWF, respectively. An interim analysis (66 subjects who had completed 20 exposure days) of human-cl rhFVIII in previously untreated patients with severe hemophilia demonstrated a cumulative incidence of 12.8% (95% confidence interval [CI], 4.5-21.2-) for high-titer inhibitors and 20.8% (95% CI, 10.7-31) for overall inhibitors.20 Of note, these rates were seen in a population that is historically at lower risk of inhibitors. These sorts of differences make comparisons across studies hazardous and the generalizability of the results unclear. Given the immunomodulatory and tolerogenic properties of the Fc fragment of immunoglobulin (Ig), it is hypothesized that rFVIIIFc could improve tolerance to FVIII. Retrospective data with rFVIIIFc suggests that, when used in subjects with a high risk for ITI failure, there is a rapid decline in inhibitor titer and time to tolerization, as well as efficacy, in rescue ITI in those who have failed other attempts.21
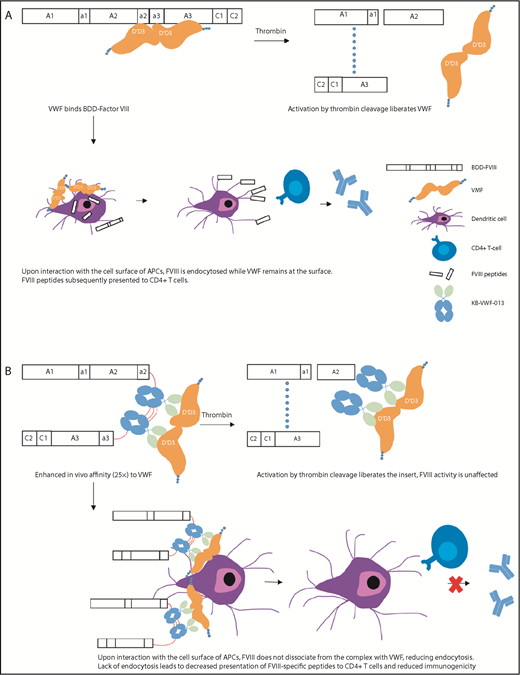
Activation and immune processing mechanisms of FVIII-KB013bv compared with BDD-FVIII. (A) BDD-FVIII binds VWF. Following activation by thrombin, FVIII is activated to FVIIIa and dissociates from VWF. Alternately, the complex circulates and interacts with the cell surface of APCs. During this interaction, VWF remains on the APC surface while FVIII is endocytosed. FVIII peptides are processed by the APC and subsequently presented to CD4+ T cells. (B) FVIII-KB013bv, an FVIII-nanobody fusion protein, replaces B-domain residues with 2 copies of KB-VWF-013. KB-VWF-013 recognizes the FVIII-binding D′D3 region and binds VWF with 25-fold higher affinity compared with BDD-FVIII. Activation by thrombin cleavage liberates the nanobody insert, which likely remains attached to VWF. FVIII activity and VWF activity are unaffected. Upon interaction of the complex with APCs, FVIII does not dissociate and remains at the cell surface, reducing internalization. Lack of endocytosis leads to decreased presentation of FVIII peptides by the major histocompatibility complex class II, reducing immunogenicity.
Activation and immune processing mechanisms of FVIII-KB013bv compared with BDD-FVIII. (A) BDD-FVIII binds VWF. Following activation by thrombin, FVIII is activated to FVIIIa and dissociates from VWF. Alternately, the complex circulates and interacts with the cell surface of APCs. During this interaction, VWF remains on the APC surface while FVIII is endocytosed. FVIII peptides are processed by the APC and subsequently presented to CD4+ T cells. (B) FVIII-KB013bv, an FVIII-nanobody fusion protein, replaces B-domain residues with 2 copies of KB-VWF-013. KB-VWF-013 recognizes the FVIII-binding D′D3 region and binds VWF with 25-fold higher affinity compared with BDD-FVIII. Activation by thrombin cleavage liberates the nanobody insert, which likely remains attached to VWF. FVIII activity and VWF activity are unaffected. Upon interaction of the complex with APCs, FVIII does not dissociate and remains at the cell surface, reducing internalization. Lack of endocytosis leads to decreased presentation of FVIII peptides by the major histocompatibility complex class II, reducing immunogenicity.
Novel FIX products
As a result of the success of bioengineering efforts with rFIX, EHL FIX products have achieved 2.4- to 4.8-fold extension over conventional products22-24 and allow dosing every 1 to 2 weeks for prophylaxis. BIVV002, a novel rFIXFc fusion protein uses recombinant XTEN conjugates to improve in vivo efficacy via subcutaneous injection compared with IV dosing in HB mice.25 FIX variant CB2679d is an rFIX with enhanced properties, including resistance to antithrombin (AT) inhibition and increased affinity for FVIIIa. It exhibits 22-fold enhanced potency in vitro and in in vivo animal models, with 8-fold prolongation of plasma activity.26 It was evaluated in a phase 1/2 trial in severe HB subjects with a cross-over comparison of IV administration compared with rFIX, followed by an IV to subcutaneous ascending-dose cohort and then a multidose subcutaneous cohort.27 In the highest tier of the multidose subcutaneous dosing cohort, 2 subjects were given a loading dose of 75 IU/kg IV, followed by 9 daily subcutaneous doses of 150 IU/kg. Plasma FIX levels were maintained at >20%, and the 2 subjects achieved trough FIX levels of 34% and 31%. Unfortunately, 1 subject showed the presence of a nonneutralizing antibody and a neutralizing antibody was identified in a second subject.
Ultimately, a noninvasive treatment option would provide further benefit in terms of ease of administration, lower cost, and improved compliance (Figure 2). Two main challenges exist with regard to oral delivery of coagulation proteins: functional preservation and bioavailability. The protein must pass through acidity and proteolytic enzymes in the stomach, maintain its structure, and be absorbed in the small intestine.28 pH-responsive anionic complexation hydrogels exploit the physiologic differences between stomach and small intestine. In the low-pH environment, hydrogen bonding between etheric oxygen of PEG chains and the methylacrylic acid carboxyl group results in a collapsed polymer network and smaller mesh size. As the pH increases, deprotonation disrupts the complex and causes electrostatic repulsion of the methylacrylic acid groups, swelling of the hydrogel, and increased mesh size, resulting in protein delivery.29 Further incorporation of a cross-linking agent, specifically designed for degradation by trypsin, increases site-specific release of FIX in the small intestine.30 Although challenges to bioavailability with this approach may limit its utility for prophylaxis, it may be effective for oral tolerization. FIX can also be fused with a transmucosal carrier (cholera toxin B subunit [CTB]), allowing for production within commercial lettuce using chloroplast vectors.31 The CTB FIX is stable in lyophilized lettuce cells. When the lettuce cells were fed to HB mice, they induced latency-associated peptide+ regulatory T cells and suppressed inhibitor/IgE formation and anaphylaxis against FIX. This observation was confirmed in an HB dog model in which the lyophilized CTB FIX–expressing lettuce cells led to robust suppression of IgG/inhibitor and IgE formation against IV-administered FIX in 3 of 4 dogs, which was accompanied by marked shortening of coagulation times by IV FIX in the orally tolerized dogs.32

Expanded modes of administration with novel therapeutics for hemophilia. Hemophilia therapeutics have traditionally been limited to IV delivery, but new technologies have expanded to allow additional modes of administration. Subcutaneous injection of bioengineered clotting factors (N8-GP, BIVV002, CB2679d), as well as novel nonfactor therapeutics, may reduce the treatment burden, particularly in those patients with difficult IV access. Oral administration (CTB-FIX and pH-responsive anionic complexation hydrogels) would provide a noninvasive option, improving ease of administration, as well as patient compliance.
Expanded modes of administration with novel therapeutics for hemophilia. Hemophilia therapeutics have traditionally been limited to IV delivery, but new technologies have expanded to allow additional modes of administration. Subcutaneous injection of bioengineered clotting factors (N8-GP, BIVV002, CB2679d), as well as novel nonfactor therapeutics, may reduce the treatment burden, particularly in those patients with difficult IV access. Oral administration (CTB-FIX and pH-responsive anionic complexation hydrogels) would provide a noninvasive option, improving ease of administration, as well as patient compliance.
A paradigm shift: from peaks and troughs to steady-state hemostatic correction
Successful prophylaxis depends on maintaining threshold levels of factor in the plasma. Data from prospective clinical trials have shown that the amount of time per week spent with an FVIII trough level < 1% is directly correlated with bleeding risk.33 This had been the hypothesis that drove the pioneering prophylaxis strategies in Sweden, with the intent of converting the severe hemophilia bleeding phenotype to a moderate phenotype.3 Based on these insights, there has been new interest in elucidating individual PK profiling to guide a personalized optimal prophylaxis regimen. The target trough level of ≥1% residual activity is arbitrary, however, because this threshold does not prevent all bleeding nor does falling below this level inevitably result in bleeding. Several additional factors may contribute to the need to maintain higher trough levels to optimize clinical outcomes: active lifestyle, underlying joint synovitis, or arthropathy. Accordingly, peak levels, area under the curve, and time spent above X% (where X may vary by individual) are all interrelated and may be critical to preventing bleeding, particularly during peak physical activities or for protection from unexpected trauma.
A post hoc analysis of data from a prospective study of PK-guided FVIII prophylaxis given every third day demonstrated that higher peak levels, higher area under the curve, and more time spent per week with FVIII levels > 20% provided increased protection from joint and nonjoint bleeding.34 This analysis predicted that a target trough level of ≥5% would have led to ∼71% of subjects achieving no spontaneous bleeds. This is being realized with some EHL factor products. Although EHL factor products can reduce the frequency of IV dosing for effective prophylaxis, the higher trough levels that can be achieved, particularly with the EHL FIX products, have been associated with resolution of target joint bleeding.23,35,36 These observations highlight that there is opportunity to improve on current prophylactic strategies by aiming for higher trough levels or through alternative strategies that achieve more steady-state hemostasis via alternative mechanisms of action. This is now being achieved with novel substitution therapy and molecules that are aimed at rebalancing the hemostatic system by targeting the natural anticoagulant pathways (Figure 3).

Mechanisms of action of novel nonfactor therapeutics for hemophilia. Hemostatic nonfactor agents in varying phases of development include substitution therapies (emicizumab, BS027125) for FVIII that can restore factor Xa generation and rebalancing therapies, which knock down or disrupt the natural anticoagulants (small interfering RNA to AT and PS, monoclonal antibodies to protein C and TFPI, and an APC-specific serpin), to augment hemostasis.
Mechanisms of action of novel nonfactor therapeutics for hemophilia. Hemostatic nonfactor agents in varying phases of development include substitution therapies (emicizumab, BS027125) for FVIII that can restore factor Xa generation and rebalancing therapies, which knock down or disrupt the natural anticoagulants (small interfering RNA to AT and PS, monoclonal antibodies to protein C and TFPI, and an APC-specific serpin), to augment hemostasis.
Substitution therapy
Emicizumab, a humanized bispecific monoclonal antibody that binds and bridges FX and FIXa, was developed to restore the missing function of FVIIIa.37 It has no structural homology to FVIII and, thus, would not be expected to induce inhibitors to FVIII or be inhibited by existing FVIII inhibitors. It carries the additional advantages of a subcutaneous mode of administration and long half-life (∼30 days).38 PK modeling showed that, after a weekly loading dose, subjects could be maintained at a steady-state plasma level with weekly, biweekly, or monthly dosing that would be sufficient to maintain the majority of subjects bleed-free, as tested in the clinical trial program39 (Table 1). HAVEN 1 was a phase 3 study in severe HA with inhibitors (HAwIs) patients that demonstrated safety and efficacy at set dosing administered weekly, an 87% reduction in annualized bleeding rate (ABR) compared with no prophylaxis, and a 79% reduction compared with prior BPA prophylaxis (obtained within a noninterventional study).40 Within this study, 63% of subjects experienced zero treated bleeds (≥24 weeks of follow-up) compared with only 6% without any prophylaxis. HAVEN 2 was a phase 3 study in pediatric patients with severe HAwIs on weekly dosing. Of 57 patients analyzed, 87% had no treated bleeds, and a 99% reduction in ABR was seen compared with prior BPA prophylaxis.41,42 Emicizumab prophylaxis in HA without inhibitors (HAwoIs) was evaluated in HAVEN 3 using weekly or biweekly dosing and demonstrated a 68% reduction in treated bleeds compared with prior FVIII prophylaxis.43 This is a notable observation and supports the hypothesis that the level of steady-state maintenance of hemostasis achieved with emicizumab prophylaxis can result in superior efficacy to the peaks and troughs of traditional prophylaxis. Improved adherence and the absence of significant impact of a late or missed dose also likely contributed to the improved efficacy. Impressive responses were also observed with monthly dosing in HAVEN 4, with a median ABR of 0.0 in patients with HAwIs and HAwoIs (36 of the 41 subjects) and 100% of patients preferring emicizumab to their prior treatment.43 The efficacy for the monthly regimen is consistent with that predicted from the PK modeling, in which mean trough levels of emicizumab were expected to be only marginally lower than those obtained with weekly dosing.
It has been postulated that establishing a bioequivalence for emicizumab plasma concentration with FVIII activity assays would allow for better benchmarking of the level of hemostatic control that can be expected. However, emicizumab is intrinsically very different from FVIII and lacks many of the same regulatory mechanisms for its action.44 This unique mechanism of action is likely the root cause of the thrombotic microangiopathy and other thrombotic events observed in the HAVEN 1 clinical trial program coincident with the use of higher doses of activated prothrombin complex concentrates to treat breakthrough bleeding.38 The differences between emicizumab and FVIII also compromise the ability to assign and interpret FVIII bioequivalence. There have been studies attempting to assign “FVIII-like” activity to emicizumab using activated partial thromboplastin time,37 modified clot waveform analysis,45 chromogenic FVIII utilizing human reagents,37 thrombin generation,46 and whole-blood clotting assays, such as thromboelastography and rotational thromboelastometry.47 However, these assays have been shown to be too sensitive and lack parallelism across a range of concentrations, and attempts to correlate the activity readouts with an in vivo clinical response have been inconsistent. Nevertheless, during the preclinical development, the target dosing of emicizumab was based on correlations between porcine FVIII and emicizumab in an acquired HA model in nonhuman primates.40,48 Similar correlations have not yet been established from the human experience. Yet the need for a bioassay that reports an activity measurement from emicizumab is important because anti-drug antibodies directed against emicizumab have been described, including neutralizing anti-drug antibodies that result in loss of efficacy.
Despite its demonstrated clinical efficacy, the biochemical potency of emicizumab is still limited,48 perhaps due to its binding to FIX (which may sequester it from FIXa) and FXa (which may delay FXa binding to FVa and prothrombin).46 Thus, there is an opportunity for continued innovation utilizing novel bispecific antibodies. Another variation of the bispecific antibody approach (BS027125) has been presented that is bioengineered to mimic the selectivity of FVIIIa in bridging FIXa and FX. Although it exhibits specificity to FX, substantial binding to FIX still remains.49
Hemostatic rebalancing therapies
The hemostatic system is highly regulated to ensure that coagulation mechanisms can be activated when there is injury but not unnecessarily activated when at homeostasis. With a coagulation factor deficiency, such as hemophilia, the delicate balance is tipped toward bleeding. In contrast, derangements in the natural anticoagulant pathways can lead to thrombosis. Evidence from laboratory and clinical experience demonstrates that targeting these natural anticoagulant pathways can restore the hemostatic equilibrium in the presence of a bleeding disorder.50-54 There are ongoing clinical development programs targeting each of the natural anticoagulant pathways for the extrinsic, intrinsic, and common pathways of coagulation (Table 2).
AT targets the common pathway factors Xa and thrombin. Fitusiran is a small interfering RNA (siRNA) that acts by targeting and binding AT messenger RNA in the liver, causing interference with translation and blocking synthesis.55 It is given subcutaneously and can be used in HA and HB, with and without inhibitors. Phase 1 results demonstrated dose-dependent decreases in AT of 61% at the highest weekly dose and 89% at the highest monthly dose, which correlated with increased thrombin generation in HA and HB patients without inhibitors.56 A phase 2 open label extension study followed using monthly fixed dosing in HA and HB patients with and without inhibitors.57 A median of 13 months after initial dosing, patients given both doses experienced an almost 80% reduction in AT levels. ABR was 1 in patients without inhibitors and 0 in patients with inhibitors in a post hoc analysis, although these data are limited because the dispersion was not reported. Unfortunately, an HA patient in the open label extension study suffered a cerebral sinus vein thrombosis following FVIII therapy for breakthrough bleeding that was ultimately fatal. Following a clinical hold on the trial, a risk-mitigation strategy was developed, including protocol guidelines and additional education regarding the need to use markedly reduced doses of replacement factor products and BPAs in breakthrough bleed treatment. Following these guidelines, there have been no further thromboembolic events reported within the clinical trial program.
Tissue factor pathway inhibitor (TFPI) acts via inhibition of the extrinsic pathway. The α isoform is responsible for inhibition of prothrombinase via a K1 domain that inhibits FVIIa, a K2 domain that inhibits FXa, and a K3 domain that binds protein S (PS). Inhibition of TFPI has been shown to reduce bleeding in multiple hemophilia animal models,58-60 and knockout within hematopoietic stem cells protects hemophilia mice from bleeding.61 Multiple strategies have been attempted to inhibit TFPI, including aptamers,58 fucoidan,60 monoclonal antibodies,59 and peptide agents.62 Concizumab, a humanized monoclonal antibody against TFPI, is the furthest along in development. With a high affinity for the K2 domain, it inhibits FXa binding and prevents inhibition of the TF–FVIIa complex. Phase I data demonstrated a reduction in plasma concentrations of TFPI, functional activity for ≥14 days, improved thrombin generation in healthy volunteers, and no safety concerns.63,64 Two phase 2 trials are ongoing in HA and HB patients with and without inhibitors. Two other products (PF-06741086 in phase 2 and BAY1093884 in phase 1) are also in development and bind the K1 and K2 domains, inhibiting TFPI binding to FVIIa or FXa.
Activated protein C (APC) is a serine protease that can bind the endothelial protein C receptor, which approximates it to thrombin for its activation, whereby it functions to downregulate the amplification of FXa generation and, in turn, thrombin generation via the intrinsic pathway, through proteolysis of FVa and FVIIIa. In addition to its anticoagulant actions, APC provides cytoprotection through antiapoptotic effects, anti-inflammatory effects, and protection of the endothelial barrier through cleavage of PAR1. APC is regulated by serpins, protein C inhibitor (PCI), and α1-antitrypsin (α1AT), which are inefficient and nonspecific. To generate a more specific inhibitor, mutations were introduced in and around the scissile bond P1-P1′, the main determinant of specificity. Mutated PCI resulted in increased selectivity but a low rate of inhibition, so the serpin scaffold was changed to α1AT Pittsburgh, which rapidly inhibits APC, resulting in the candidate serpin, KRK α1AT or SerpinPC.65 Other advantages of the α1AT scaffold are its 5- to 7-day half-life, high bioavailability supporting subcutaneous mode of delivery, and low immunogenicity given high levels of endogenous α1AT. SerpinPC was found to rescue hemostasis in an HA mouse model. Notably, evidence supports that the signaling and anticoagulant functions of APC are in spatially and kinetically distinct compartments.66 The kinetics of the SerpinPC interaction with APC should allow for inhibition of the anticoagulant activity with preservation of the anti-inflammatory and cytoprotective roles. APC has also been targeted using a monoclonal antibody (HAPC1573) that specifically binds to APC but not zymogen protein C. It has been shown to protect FVa and FVIIIa from inactivation, reduce the activated partial thromboplastin time of hemophilic plasma, and enhance thrombin generation. Of particular interest, it inhibits the anticoagulant functions of APC without any effect on its cytoprotective functions when measured by histone-mediated cytotoxicity assays. Efficacy and safety have been demonstrated in monkeys, with restoration of hemostasis in a dose-dependent manner.67
The least developed target of hemostatic rebalancing therapy is inhibition of PS. PS acts as a cofactor to APC for the inactivation of FVa and FVIIIa and to TFPI for inhibition of FXa. To evaluate whether knockdown of PS would result in bleeding amelioration in HA and HB, PROS1 deficiency and PS inhibition were evaluated in hemophilic mice and demonstrated improvement in tail-bleeding assays and protection against hemarthrosis. The effect of PS inhibition was also evaluated in human HA plasma and revealed twofold to fourfold increased endogenous thrombin potential. These studies used a murine PS siRNA that has the advantages of a long half-life and possible subcutaneous administration.68 A critical qualification of this early work is known species differences. In human plasma, PS is present as free protein and in complex with C4b-binding protein, a complement regulator. Because mice do not have a C4b-binding protein–PS complex, it remains to be seen how silencing PS may affect the complement system. Additionally, PS has been shown to be a ligand and activator of TAM receptors that regulate the immune response and stimulate phagocytosis of apoptotic cells. This may lead to unintended effects of PS inhibition.69
Conclusions
Five decades of advances have brought the widespread availability of safe and effective hemophilia treatment. Yet treatment challenges remain that have prevented prophylactic therapy from achieving no breakthrough bleeding events and eliminating progressive joint deterioration in all patients (Table 3). The EHL factors have benefits that can help to overcome some challenges. However, the therapies described in this review that are not based on replacement of the missing clotting factor, such as substitution therapy and hemostatic rebalancing therapies, have shown great promise. These approaches, which do not expose the patient to FVIII or FIX, would not trigger inhibitors and would allow treatment, despite existing inhibitors.
There are other important treatment gaps that may be filled by a broader application of emicizumab or rebalancing therapies than has been investigated in the clinical trial programs to date. Current approaches to prophylaxis initiation in HA are such that infants are likely not on prophylactic therapy until the end of the first year of life or later. This is pragmatic in that spontaneous bleeding is less common, and regular IV access for prophylaxis is challenging. Yet, infants remain at risk for significant bleeding with trauma, including central nervous system hemorrhage. Emicizumab or rebalancing therapy prophylaxis could be initiated soon after birth to fill this important treatment gap. Similarly, those with moderate and mild hemophilia are not typically prescribed prophylaxis given the lower rate of bleeding and the burden of regular IV prophylaxis. Yet, serious bleeding complications and joint disease still occur within these phenotypes and would benefit from a steady-state hemostatic correction. The immunologic reaction to replacement of the missing clotting factor, particularly to FVIII, is the most serious complication of the replacement therapy approach. The substitution and hemostatic rebalancing therapies offer the potential to avoid exposure to the immunogenic proteins or to explore more tolerogenic approaches to introduction of the clotting factors once they are no longer required for hemostatic control. Ultimately, gene therapy may provide durable expression of efficacious levels of FVIII and FIX. However, that approach has not been available to pediatric patients and those with inhibitors. Preexisting immunity to viral vectors may preclude many from benefitting from this approach. Known and unknown risks related to immunologic responses, cellular stress induction, and genome integration remain. Thus, continued innovation toward alternative treatment approaches are likely to continue to advance and hopefully provide additional highly effective therapeutic opportunities for years to come.
Authorship
Contribution: A.C.W. and S.W.P. contributed equally to the concept, writing, and editing of this review.
Conflict-of-interest disclosure: A.C.W. has served as a consultant for Shire and Kedrion Biopharma. S.W.P. has received research funding from Shire and Pfizer; serves on clinical trial steering committees for BioMarin Pharamceutical, uniQure, and Bayer; and is a consultant for Shire, Pfizer, BioMarin Pharmaceutical, uniQure, Bayer, Roche/Genentech, CSL Behring, Alnylam Pharmaceuticals, Novo Nordisk, HEMA Biologics, Bioverativ, Catalyst Biosciences, DNARx, and Spark Therapeutics.
Correspondence: Steven W. Pipe, Departments of Pediatrics and Pathology, University of Michigan, 1500 E Medical Center Dr, MPB D4202, Ann Arbor, MI 48109-5718; e-mail: ummdswp@med.umich.edu.

