

TO THE EDITOR:
Ibrutinib is approved for all lines of therapy in patients with chronic lymphocytic leukemia (CLL) in the United States. Randomized studies have consistently demonstrated superior progression-free survival (PFS) in relapsed/refractory1,2 and treatment-naïve CLL patients treated with ibrutinib.3-5 Furthermore, single-agent ibrutinib improved overall survival (OS) in 2 phase 3 studies compared with ofatumumab in relapsed/refractory CLL (RESONATE)1 and with chlorambucil in treatment-naïve patients older than 65 years (RESONATE-2).3 In these studies, ibrutinib was dosed continuously at 420 mg once daily. A retrospective analysis of the RESONATE trial reported that mean ibrutinib dose intensity <95% was associated with inferior PFS but not OS.6 However, observation time of this study was limited to the first 9 months of therapy, when dose adherence is particularly important and early interruptions may indicate more advanced disease or concurrent health issues that interfere with effective delivery of therapy. In contrast, CLL progression, which tends to occur later, may have been underrepresented in this short follow-up. Similarly, a study by the UK CLL Forum reported treatment breaks >14 days within the first year of ibrutinib were associated with inferior OS.7 However, the latter study noted patients with early treatment breaks had a poor performance status and were 4 times more likely to permanently stop ibrutinib within the first year, suggesting host-related factors as a confounder. In clinical practice, ibrutinib dose modifications are often medically indicated, either to mitigate clinically significant adverse events (grade ≥3, including serious adverse events requiring hospitalizations or urgent medical interventions) or as precautionary drug holds for invasive procedures. Therefore, the question of the degree to which ibrutinib dose interruptions may limit long-term benefit deserves further study.
We analyzed a cohort of 84 treatment-naïve (n = 52) or relapsed/refractory (n = 32) CLL patients treated with ibrutinib in a phase 2 study (NCT01500733). Demographics and safety and efficacy outcomes at 5 years were previously reported.8 Eligible patients either had a TP53 aberration, defined by detection of TP53 mutation or deletion 17p (n = 53), or were age ≥65 years (n = 31). Decisions on permanent dose reductions were exclusively made and documented by the investigators. Each dose reduction event was also confirmed on a review of electronic pharmacy orders for ibrutinib at the time of analysis. Dose interruption data were based on documented patient history. Specifically, ibrutinib was held for elective procedures and at the time of serious adverse events or grade ≥3 adverse events, consistent with holding guidelines from the current US prescribing information for ibrutinib.9 To screen for noncompliance, we dispensed up to 3 months supply of ibrutinib and obtained verbal reports from patients on any missed doses at each clinic visit. The study did not use patient diaries or pill counts. Assuming ibrutinib dose reductions or interruptions occur randomly during the course of a study, patients receiving long-term therapy will accumulate more treatment breaks, resulting in a confounder, called guarantee-time bias, that interferes with conventional survival analyses.10,11 Therefore, we used both landmark analysis and a Cox regression model to assess the effect of ibrutinib dose intensity on PFS and OS.10,12 PFS and OS were estimated by the Kaplan-Meier method and compared between subgroups by the log-rank test. Statistical analyses were conducted using R software (version 3.5.1; R Foundation for Statistical Computing).
At a median follow-up of 5.1 years, 75 patients (89.3%) had missed at least 1 dose of ibrutinib, and 12 patients (14.3%) required permanent dose reductions, 10 to 280 mg per day and 2 to 140 mg per day (Figure 1A). The most common reason for treatment breaks were elective procedures (152 [45.8%] of a total 332 dose interruption events) followed by adverse events (70 [21.1%] events) and noncompliance (68 [20.5%] events). Fifty-seven patients (67.9%) missed ibrutinib for ≥8 consecutive days, and 40 (47.6%) missed ibrutinib for ≥15 consecutive days (Figure 1B). Among 12 patients who required permanent dose reductions, atrial fibrillation, in 5 patients, was the most common reason.
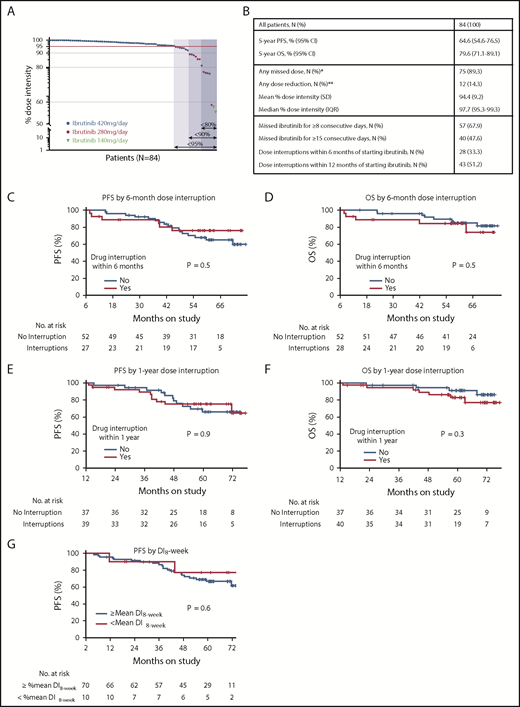
Ibrutinib dose intensity and clinical outcomes. (A) Ibrutinib dose intensity of 84 CLL patients on study. Arrows indicate subgroups defined by dose intensity (%). The darkest blue shade indicates 7 patients who had <80% dose intensity; the intermediate blue shade indicates those with <90% dose intensity; the lightest shade indicates those with <95% dose intensity. Patients receiving full-dose ibrutinib (420 mg per day) are represented by black circles; red circles indicate patients who had permanent dose reductions to 280 mg per day; purple triangles indicate reductions to 140 mg per day. (B) Summary of the study cohort and subgroups divided by dose intensity. (C-F) Landmark analysis of PFS (C,E) and OS (D,F) of subgroups divided by ibrutinib interruption of any duration within the first 6 months (C,D) or 1 year (E,F) of starting ibrutinib. The number at risk for 6-month dose adherence was 79 for PFS and 80 for OS, with an additional patient who progressed and was alive at 6 months included in the OS analysis. The numbers at risk for 1-year dose adherence were 76 and 77 for PFS and OS, respectively. (G) A landmark analysis of PFS of subgroups divided by 8-week dose intensity (DI8-week) above vs at or below mean of the cohort. *The most common reasons for dose interruption were elective procedures (ie, Mohs surgery, ophthalmologic procedures, arthroscopy or intraarticular injections, dental procedures) followed by toxicity and noncompliance. **The most common reason for permanent dose reduction was atrial fibrillation (41.7%) followed by arthralgia (25%) and diarrhea (16.7%). IQR, interquartile range; SD, standard deviation.
Ibrutinib dose intensity and clinical outcomes. (A) Ibrutinib dose intensity of 84 CLL patients on study. Arrows indicate subgroups defined by dose intensity (%). The darkest blue shade indicates 7 patients who had <80% dose intensity; the intermediate blue shade indicates those with <90% dose intensity; the lightest shade indicates those with <95% dose intensity. Patients receiving full-dose ibrutinib (420 mg per day) are represented by black circles; red circles indicate patients who had permanent dose reductions to 280 mg per day; purple triangles indicate reductions to 140 mg per day. (B) Summary of the study cohort and subgroups divided by dose intensity. (C-F) Landmark analysis of PFS (C,E) and OS (D,F) of subgroups divided by ibrutinib interruption of any duration within the first 6 months (C,D) or 1 year (E,F) of starting ibrutinib. The number at risk for 6-month dose adherence was 79 for PFS and 80 for OS, with an additional patient who progressed and was alive at 6 months included in the OS analysis. The numbers at risk for 1-year dose adherence were 76 and 77 for PFS and OS, respectively. (G) A landmark analysis of PFS of subgroups divided by 8-week dose intensity (DI8-week) above vs at or below mean of the cohort. *The most common reasons for dose interruption were elective procedures (ie, Mohs surgery, ophthalmologic procedures, arthroscopy or intraarticular injections, dental procedures) followed by toxicity and noncompliance. **The most common reason for permanent dose reduction was atrial fibrillation (41.7%) followed by arthralgia (25%) and diarrhea (16.7%). IQR, interquartile range; SD, standard deviation.
To cumulatively capture dose intensity, we integrated both dose reductions and interruptions as a fraction of the target ibrutinib dose of 420 mg per day, using methods previously described.6 For patients who stopped ibrutinib for reasons other than disease progression, dose intensity was calculated up to the off-study time point. The mean dose intensity in our study was 94.4% (range, 55.3% to 100%), similar to that in the RESONATE trial.6 Dose intensity of <95% was observed in 21 patients (25.0%), <90% in 14 patients (16.7%), and <80% in 7 patients (8.3%). We observed a wide variation in time from start of ibrutinib to first dose interruption (2 days to 43 months).
Twenty-three (27.4%) of 84 patients progressed, including 5 with Richter’s transformation; 18 patients died, including 13 as a result of disease progression. The mean dose intensity of the patients who progressed was 97.7% (range, 88.4% to 100%). Five-year PFS and OS estimates for the whole cohort were 64.6% (95% confidence interval [CI], 54.6% to 76.5%) and 79.6% (95% CI, 71.1% to 89.1%), respectively. Five-year PFS and OS estimates in patients who missed ibrutinib for ≥8 consecutive days were 72.1% (95% CI, 60.7% to 85.7%) and 90.1% (95% CI, 82.2% to 98.8%), respectively. Twenty-eight patients (33.3%) had early treatment interruptions of any duration within 6 months of starting ibrutinib, and 43 patients (51.2%) had interruptions within the first year. Landmark analyses at 6 months and 1 year after starting ibrutinib showed early treatment breaks did not affect PFS or OS (Figure 1C-F). Additionally, we assessed dose intensity at 8 weeks using methods previously described.6 Four patients who progressed or died before reaching 8 weeks on study were excluded from this analysis, leaving 80 evaluable patients. The mean dose intensity at 8 weeks of the overall study population was 98.6%. Ten patients had early dose interruptions within 8 weeks, and their dose intensity at 8 weeks was below the mean. Landmark analysis at 8 weeks demonstrated no difference in PFS between subgroups of patients whose dose intensity at 8 weeks was below the mean vs those whose dose intensity at 8 weeks was at or above the mean (Figure 1G). Our experience with permanent dose reductions is limited. However, we note that with a median follow-up of 39.4 months from the first dose reduction, only 1 (8.3%) of 12 patients progressed.
To adjust for dosing changes during the treatment course, we included dose intensity as a time-dependent covariate in the Cox regression model. Subgroups defined by various dose intensity cutoffs (95%, 90%, and 80%) had comparable PFS (hazard ratio [HR] per 1% decrease in dose intensity, 0.95; 95% CI, 0.86-1.04; P = .26) and OS (HR, 0.97; 95% CI, 0.88-1.06; P = .48). In multivariate analysis, dose intensity was not associated with PFS or OS (both P > .3).
In conclusion, we found no evidence that clinically indicated ibrutinib dose reductions or interruptions compromised long-term outcomes. Dose intensity was high in our study (94.4%). In general practice, dose intensity could be lower and might affect outcome. Our findings should not be extended to general noncompliance with standard ibrutinib therapy. Although our study is limited in size, because of long-term follow-up, we captured more progression and dose reduction events than Barr et al6 included in their report. Our conclusions, in part, differ from those reached by Barr et al. However, we agree that maintaining high dose intensity is important, and we avoided any unnecessary dose interruptions throughout the study, particularly in the first 6 months of therapy.
There are notable differences between our study and the RESONATE trial. Our study population was enriched in high-risk disease characteristics, and these patients would be expected to strictly adhere to long-term therapy, which was reflected in a mean dose intensity of 94.4% even at 5-year median follow-up. Our study was enriched with previously untreated patients, which likely improved long-term outcomes compared with patients with relapsed/refractory CLL enrolled in the RESONATE trial. In the RESONATE trial, most progression events and deaths occurred in the first 6 months, suggesting that disease-related factors may have contributed to inferior clinical outcomes. In contrast, most progression events in our study occurred after the first year of therapy, and differences in dose intensity in the first 6 or 12 months did not translate into differences in outcome. Furthermore, progression events in the RESONATE trial were called by an independent review committee, and some of these events were thought to be related to dose interruptions and transient in nature, as evidenced by 11 patients (42%) continuing therapy without clinical progression. In our study, we considered drug holds as a confounder for response assessments and required evidence of sustained progression of disease once therapy was resumed.
Our observation is consistent with known mechanisms of secondary resistance to ibrutinib, which is most commonly caused by clonal evolution of CLL cells carrying BTK or PLCG2 mutations that often manifest only after years of therapy.13-15 Therefore, short-term interruptions of ibrutinib are unlikely to contribute to the emergence of drug-resistant clones. Rather, treatment history and the biologic and genetic make-up of CLL at the start of therapy, reflected in TP53 aberrations,14,16 complex karyotype,14,17 and CD49d expression,18 predict long-term outcomes with ibrutinib.15-17 In addition to biologic principles, experience in other clinical studies supports the conclusion that clinically indicated dose reductions do not translate into inferior outcomes.6,7,19,20
Acknowledgments
The authors thank the patients who participated in this trial and their families, as well as Ovsanna Melikyan and Adriana Byrnes for protocol support and Manuk Manukyan, Larisa Bezkorovaynaya, and Tatyana Sarkisova for data management.
This research was supported by the Intramural Research Program of the National, Heart, Lung and Blood Institute, National Institutes of Health, and by Pharmacyclics, which provided study drug and research support for the study.
Authorship
Contribution: I.E.A. and A.W. conducted the clinical trial and designed the study concept; I.E.A., S.S., and N.B. collected and analyzed the data; X.T. performed statistical analysis; I.E.A., X.T., and A.W. wrote the manuscript; and all authors reviewed and approved the manuscript.
Conflict-of-interest disclosure: A.W. received research support from Pharmacyclics LLC, an AbbVie company. The remaining authors declare no competing financial interests.
Correspondence: Inhye E. Ahn, Hematology Branch, NHLBI, NIH, Building 10, CRC 5-5142, 10 Center Dr, Bethesda, MD 20892-1202; e-mail: inhye.ahn@nih.gov; and Adrian Wiestner, Hematology Branch, NHLBI, NIH, Building 10, CRC 3-5140, 10 Center Dr, Bethesda, MD 20892-1202; e-mail: wiestnera@mail.nih.gov.

