Abstract
Primary cutaneous lymphomas are a heterogeneous group of T- and B-cell lymphomas that present in the skin with no evidence of extracutaneous disease at the time of diagnosis. The 2005 World Health Organization–European Organization for Research and Treatment of Cancer (WHO-EORTC) consensus classification has served as a golden standard for the diagnosis and classification of these conditions. In September 2018, an updated version of the WHO-EORTC was published in the fourth edition of the WHO Classification of Skin Tumours Blue Book. In this classification, primary cutaneous acral CD8+ T-cell lymphoma and Epstein-Barr virus positive (EBV+) mucocutaneous ulcer are included as new provisional entities, and a new section on cutaneous forms of chronic active EBV disease has been added. The term “primary cutaneous CD4+ small/medium T-cell lymphoma” was modified to “primary cutaneous CD4+ small/medium T-cell lymphoproliferative disorder” because of its indolent clinical behavior and uncertain malignant potential. Modifications have also been made in the sections on lymphomatoid papulosis, increasing the spectrum of histologic and genetic types, and primary cutaneous marginal zone lymphomas recognizing 2 different subtypes. Herein, the characteristic features of these new and modified entities as well as the results of recent molecular studies with diagnostic, prognostic, and/or therapeutic significance for the different types of primary cutaneous lymphomas are reviewed. An update of the frequency and survival of the different types of primary cutaneous lymphomas is provided.
Medscape Continuing Medical Education online
![]()
In support of improving patient care, this activity has been planned and implemented by Medscape, LLC and the American Society of Hematology. Medscape, LLC is jointly accredited by the Accreditation Council for Continuing Medical Education (ACCME), the Accreditation Council for Pharmacy Education (ACPE), and the American Nurses Credentialing Center (ANCC), to provide continuing education for the healthcare team.
Medscape, LLC designates this Journal-based CME activity for a maximum of 1.0 AMA PRA Category 1 Credit(s)™. Physicians should claim only the credit commensurate with the extent of their participation in the activity.
Successful completion of this CME activity, which includes participation in the evaluation component, enables the participant to earn up to 1.0 MOC points in the American Board of Internal Medicine's (ABIM) Maintenance of Certification (MOC) program. Participants will earn MOC points equivalent to the amount of CME credits claimed for the activity. It is the CME activity provider's responsibility to submit participant completion information to ACCME for the purpose of granting ABIM MOC credit.
All other clinicians completing this activity will be issued a certificate of participation. To participate in this journal CME activity: (1) review the learning objectives and author disclosures; (2) study the education content; (3) take the post-test with a 75% minimum passing score and complete the evaluation at http://www.medscape.org/journal/blood; and (4) view/print certificate. For CME questions, see page 1791.
Disclosures
Associate Editor Laurie H. Sehn served as an advisor or consultant for AbbVie, Amgen, Celgene, Janssen Pharmaceuticals, Karyopharm Therapeutics, Merck & Co., Roche/Genentech, Seattle Genetics, and TG Therapeutics and received grants for clinical research from Roche/Genentech. Author Rein Willemze served as an advisor or consultant for Takeda. Laurie Barclay, freelance writer and reviewer, Medscape, LLC and the remaining authors declare no competing financial interests.
Learning objectives
Upon completion of this activity, participants will be able to:
Describe new provisional entities now included in classification of primary cutaneous lymphomas, according to the 2018 update of the World Health Organization–European Organization for Research and Treatment of Cancer (WHO-EORTC) consensus classification
Determine modifications in the sections on lymphomatoid papulosis and primary cutaneous marginal zone lymphoma from the previous WHO-EORTC consensus classification to the 2018 update
Explain other changes in classification of primary cutaneous lymphomas from the previous WHO-EORTC consensus classification to the 2018 update
Release date: April 18, 2019; Expiration date: April 18, 2020
Primary cutaneous lymphomas are defined as non-Hodgkin lymphomas presenting in the skin with no evidence of extracutaneous disease at the time of diagnosis. Primary cutaneous lymphomas include a heterogeneous group of cutaneous T-cell lymphomas (CTCLs) and cutaneous B-cell lymphomas (CBCLs). In the Western world, CTCLs constitute ∼75% to 80% of all primary cutaneous lymphomas, and CBCLs 20% to 25%.1 These different types of CTCLs and CBCLs have highly characteristic clinical and histologic features, often a completely different clinical behavior and prognosis compared with morphologically similar nodal lymphomas that may involve the skin secondarily, and require a different type of treatment. Primary cutaneous lymphomas were therefore included as distinct entities in current lymphoma classifications. In the past decade, the World Health Organization–European Organization for Research and Treatment of Cancer (WHO-EORTC) consensus classification has served as a gold standard for the diagnosis and classification of primary cutaneous lymphomas. This classification was published in 2005 and was included in the 2006 WHO classification of Skin Tumors Blue Book.1,2 Most of that classification was subsequently incorporated in the 2008 WHO classification and its 2016 revision.3 In August 2018, an updated version of the WHO-EORTC classification was published in the fourth edition of the WHO Classification of Skin Tumors Blue Book (Table 1).4 The terminology and the definitions of the different types of primary cutaneous lymphomas in the updated WHO-EORTC classification are, for the most part, identical to those used in the 2017 WHO Hematopoietic and Lymphoid Tumor Blue Book. Compared with the 2005 WHO-EORTC classification, some new provisional entities have been added, whereas the terminology of some other conditions has been modified.
In this review, the main characteristics of the new and modified disease entities in the updated WHO-EORTC classification are described, and the results of recent molecular studies resulting in new diagnostic and prognostic biomarkers are presented. In addition, an update of the frequency and survival of the different types of primary cutaneous lymphomas, based on data included in the Dutch and Austrian cutaneous lymphoma registries between 2002 and 2017, is provided in Table 2.
MF, variants of MF, and SS
Mycosis fungoides (MF) and Sézary syndrome (SS) are the classic types of CTCL. MF is the most common type and accounts for 60% of CTCLs and almost 50% of all primary cutaneous lymphomas.1 In the WHO-EORTC classification, folliculotropic MF (FMF), pagetoid reticulosis, and granulomatous slack skin are recognized as distinct variants of MF, because of their distinctive clinicopathologic features, clinical behavior, and/or prognosis. Whereas pagetoid reticulosis and granulomatous slack skin are extremely rare, FMF accounts for ∼10% of all cases of MF.5,6 FMF differs from the classic form of MF by the presence of folliculotropic infiltrates, often with sparing of the epidermis, the preferential localization of skin lesions in the head and neck region, and the presence of (grouped) follicular papules, acneiform lesions, and associated alopecia. Previous studies emphasized that FMF is generally less responsive to several skin-directed therapies and runs a more aggressive clinical course compared with classic MF, and should therefore be treated more aggressively.5,7,8 However, recent clinicopathologic studies defined a subgroup of FMF patients with an indolent clinical behavior and an excellent prognosis, similar to that of early-stage classic MF.9,10 Recognition of indolent and more aggressive subgroups of FMF is important from a therapeutic point of view. It suggests that a stepwise, stage-adapted therapeutic approach can be followed, similar as in early and advanced stage classic MF.11
SS
SS is a rare leukemic type of CTCL, traditionally defined by the triad of pruritic erythroderma, generalized lymphadenopathy, and clonally related neoplastic T cells with cerebriform nuclei (Sézary cells) in the skin, lymph nodes, and peripheral blood. Differentiation between early-stage SS and erythrodermic inflammatory dermatoses (EIDs) may be very difficult.12 The histologic features of SS may be similar to those in MF. However, the superficial perivascular infiltrates may be sparse, epidermotropism may be minimal or absent, and in as many as one-third of biopsies from patients with otherwise classic SS, the histologic picture may be aspecific.12 Because both clinical and histopathological presentation may be nonspecific, demonstration of peripheral blood involvement is crucial for the diagnosis of SS. Criteria for blood involvement include, in addition to demonstration of clonally related neoplastic T cells in skin and peripheral blood, either an absolute Sézary cell count of >1000/µL, or an expanded CD4+ T-cell population resulting in a CD4/CD8 ratio ≥10, CD4+/CD7− cells ≥40%, or CD4+/CD26− cells ≥30%.
Recent studies have described new biomarkers, including, among others, PD-1 (CD279) and KIRDL2 (CD158k), which can facilitate differentiation between SS and EIDs both in skin and peripheral blood (Figure 1).13,14 Gene expression analyses of circulating Sézary cells showed a characteristic pattern with overexpression of PLS3, TWIST1, DNM3, EPH4, CD158k/KIRDL2, and NKp46, and reduced expression of STAT4.15,16 Combinations of these altered genes have been shown to differentiate reliably between SS and EIDs, but such diagnostic panels are not yet used in daily practice. Genetic alterations in SS are diverse and complex. Recent large-scale genomic studies showed alterations in genes involved in T-cell activation, cell-cycle regulation, DNA damage repair, chromatin remodeling, NF-κB activation, and JAK-STAT signaling.17,18 These studies have not only contributed to new insights in the molecular pathogenesis of SS, but also provided new therapeutic targets, which are currently tested in clinical trials (reviewed in Elenitoba-Johnson and Wilcox17 ).
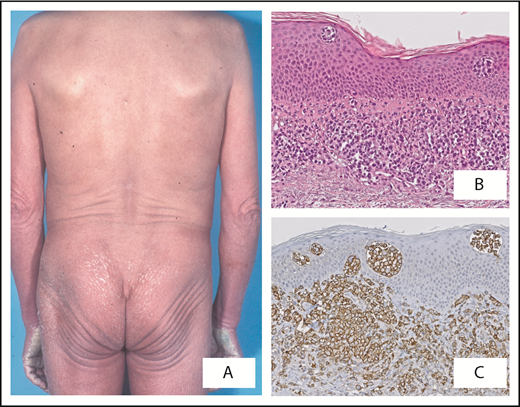
Sézary syndrome. Patient presenting with (A) erythroderma. (B) Band-like infiltrate of atypical lymphoid cells in superficial dermis with formation of intraepidermal (Pautrier) microabscesses. (C) Strong expression of CD279 (PD-1) by neoplastic T cells is a useful marker to differentiate Sézary syndrome from erythrodermic inflammatory dermatoses. Original magnification ×40 (B-C); hematoxylin and eosin (B) and immunoperoxidase (C) stain.
Sézary syndrome. Patient presenting with (A) erythroderma. (B) Band-like infiltrate of atypical lymphoid cells in superficial dermis with formation of intraepidermal (Pautrier) microabscesses. (C) Strong expression of CD279 (PD-1) by neoplastic T cells is a useful marker to differentiate Sézary syndrome from erythrodermic inflammatory dermatoses. Original magnification ×40 (B-C); hematoxylin and eosin (B) and immunoperoxidase (C) stain.
Primary cutaneous CD30+ T-cell LPD
Primary cutaneous CD30+ lymphoproliferative disorders (LPDs) are the second most common group of CTCL, accounting for ∼25% of all CTCLs.1 This group includes primary cutaneous anaplastic large lymphoma (C-ALCL) and lymphomatoid papulosis (LyP), which form a spectrum of disease.1 Because of the overlapping histologic and phenotypic features, clinical presentation and clinical course are used as decisive criteria to differentiate between LyP and C-ALCL and to select the appropriate type of treatment.19,20 C-ALCL presents as solitary, grouped, or, uncommonly, multifocal nodules and tumors. Cutaneous relapses are common, but extracutaneous dissemination occurs in only 10% to 15% of patients. LyP is characterized by a chronic course of recurrent, self-healing papulonecrotic or nodular skin lesions. The histologic picture of LyP is extremely variable and may resemble different types of CTCLs (Table 2). In addition to the 3 original subtypes (LyP types A, B, and C), the 2018 update of the WHO-EORTC classification also recognizes the more recently described types D (resembling primary cutaneous aggressive epidermotropic cytotoxic T-cell lymphoma),21 type E (angiocentric and angiodestructive and clinically characterized by large necrotic eschar-like lesions),22 a new subtype23 characterized by the presence of chromosomal rearrangements involving the DUSP-IRF4 locus on 6p25.3,24 and some even more uncommon variants.21,22,24 The frequency, predominant phenotype, and type of CTCLs these mimic are summarized in Table 2. Recognition of these different types of LyP is important to avoid misdiagnosis of other often more aggressive types of CTCLs, but has no therapeutic or prognostic implications.
ALK, DUSP22-IRF4, and TP63 rearrangements
In primary systemic ALCL, distinction is made between ALK+ and ALK− ALCLs, the former having a much better prognosis. In the group of systemic ALK− ALCLs, 2 other recurrent rearrangements have been detected, 1 involving the DUSP22-IRF4 locus on chromosome 6p25.3 and the other involving the TP63 gene on chromosome 3q28, which are associated with a favorable and a very poor prognosis, respectively.25 Unlike systemic ALCL, the vast majority of C-ALCL does not carry translocations involving the ALK gene and does not express ALK. Expression of ALK protein therefore strongly suggests secondary cutaneous involvement of a systemic ALK+ ALCL. However, unusual cases of ALK+ C-ALCLs, including those showing strong nuclear and cytoplasmic staining characteristic of the t(2;5) chromosomal translocation and cases expressing cytoplasmic ALK protein, indicative of a variant translocation, have been reported.26-29 Many of these cases had an excellent prognosis. However, rapid progression to systemic ALCL has been reported as well (Figure 2). It is at present impossible to predict whether such ALK+ cases presenting with skin lesions only will run an indolent or aggressive course. Rearrangements of the DUSP22-IRF4 locus are found in ∼25% of C-ALCL and in a small subset of LyP, but do not have prognostic significance.30 Histologically, these lesions show a biphasic growth pattern with CD30+ small cerebriform lymphocytes in the epidermis and large CD30+ transformed cells in the dermis, and show reduced expression of cytotoxic proteins.24,31,32 TP63 gene rearrangements are associated with a poor survival in ALK− systemic ALCL, but are not or are rarely found in C-ALCL.33,34 A novel recurrent NPM1-TYK2 gene fusion resulting in constitutive STAT signaling has been described in both C-ALCL and LyP.35 TYK2 breaks were found in 15% of primary cutaneous CD30+ LPDs. In contrast to tumor stage MF and peripheral T-cell lymphoma, NOS, loss of 9p21.3 harboring CDKN2A suppressor gene is rarely observed in C-ALCL.36
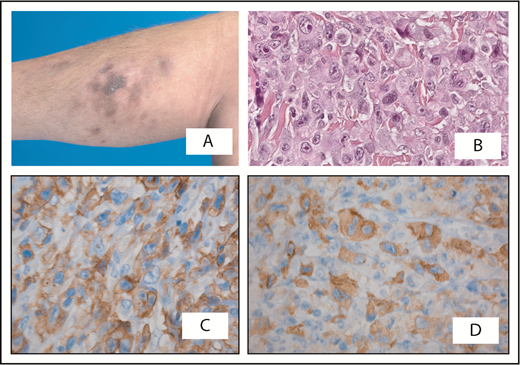
Cutaneous anaplastic large cell lymphoma presenting with multiple skin lesions on the right lower leg. (A) Part disappeared spontaneously. (B) Histologic examination shows a diffuse infiltrate of large anaplastic cells, which are positive for CD30 (C) and show cytoplasmic staining for ALK (D). Staging was negative; initially, an expectant policy was followed. Twelve months after diagnosis, the patient developed systemic disease with involvement of the lungs and bone marrow. Treatment with multiagent chemotherapy was unsuccessful and she died 18 months after diagnosis. Original magnification ×400 (B-D); hematoxylin and eosin (B) and immunoperoxidase (C-D) stain.
Cutaneous anaplastic large cell lymphoma presenting with multiple skin lesions on the right lower leg. (A) Part disappeared spontaneously. (B) Histologic examination shows a diffuse infiltrate of large anaplastic cells, which are positive for CD30 (C) and show cytoplasmic staining for ALK (D). Staging was negative; initially, an expectant policy was followed. Twelve months after diagnosis, the patient developed systemic disease with involvement of the lungs and bone marrow. Treatment with multiagent chemotherapy was unsuccessful and she died 18 months after diagnosis. Original magnification ×400 (B-D); hematoxylin and eosin (B) and immunoperoxidase (C-D) stain.
CTCLs other than MF, SS, and primary cutaneous CD30+ LPD
CTCLs other than MF, SS, and primary cutaneous CD30+ LPD are rare and together account for <10% of CTCLs.1 The clinical, histopathologic, and immunophenotypic characteristics of these other types of CTCLs are summarized in Table 3. Although primary cutaneous γ/δ T-cell lymphoma characteristically show a TCRγδ+, βF1− T-cell phenotype, expression of TCRγδ has also been found in rare cases of otherwise classic MF or LyP.37,38 Such cases have the same indolent course as cases with an αβ T-cell phenotype, and should be diagnosed as MF or LyP, irrespective of phenotype. Conditions that have been modified or have been newly added to the classification, including chronic active Epstein-Barr virus (EBV) infection in childhood, primary cutaneous acral CD8+ T-cell lymphoma, and primary cutaneous CD4+ small/medium T-cell LPD, are discussed in more detail in the following section.
Chronic active EBV infection in childhood
The updated WHO-EORTC classification contains a new section on EBV+ LPD in childhood, which includes hydroa vacciniforme-like LPD (HV-like LPD) and hypersensitivity reactions to mosquito bites. Both are cutaneous manifestations of chronic active EBV infection with a risk for progression to systemic EBV+ T- or natural killer (NK)-cell lymphoma.39 In addition, some of these cases will have evidence of systemic chronic active EBV infection. Most cases of HV-like LPD have a CD8+ T-cell phenotype, whereas hypersensitivity reactions to mosquito bites more often have an NK-cell phenotype.39 HV-like LPD is used as an encompassing term for cases previously referred to as HV and HV-like T-cell lymphoma. These disorders are seen mainly in children and adolescents from Asia or in indigenous populations from Central and South America and Mexico.40,41
Clinically, classic HV presents with a papulovesicular eruption on sun-exposed skin areas, in particular the face, the ear lobes, and the back of the hands, often with seasonal activity, but without systemic symptoms.42 In more severe cases, skin lesions are localized in sun-exposed and nonexposed skin areas, facial swelling and extensive ulceration are common, and systemic symptoms, such as fever, wasting, lymphadenopathy, and hepatosplenomegaly, may be present.43,44 Patients with mosquito bite hypersensitivity typically develop ulceronecrotic lesions at the site of the mosquito bite and may demonstrate similar systemic symptoms as seen in patients with HV-like lymphoma.40,45
The clinical course is variable and patients may have recurrent skin lesions for many years before progression to systemic lymphoma.
Primary cutaneous acral CD8+ T-cell lymphoma
Primary cutaneous acral CD8+ T-cell lymphoma is a newly described entity histologically characterized by a diffuse infiltrate of medium-sized CD8+ cytotoxic T cells, suggesting an aggressive malignant lymphoma, but with an indolent clinical behavior.46 This condition, initially designated “indolent CD8+ lymphoid proliferation of the ear,” has been included as a new provisional entity in the updated WHO-EORTC classification.
Patients typically present with a solitary, slowly progressive papule or nodule, preferentially located on the ear or less commonly on other acral sites, including the nose and the foot (Figure 3A).46,47 These lesions show a diffuse proliferation of clonal medium-sized blast cells throughout the dermis, separated from the epidermis by a clear grenz zone. The atypical cells show a CD3+, CD4−, CD8+, and CD30− T-cell phenotype with variable loss of pan-T-cell antigens (CD2, CD5, CD7). They are positive for TIA-1, but unlike other types of CD8+ CTCLs, are negative for other cytotoxic proteins (granzyme B, perforin).48 CD68 often shows a positive Golgi dot-like staining (Figure 3).49 In almost all cases, the proliferation rate is very low (<10%). EBV is negative.

Primary cutaneous acral CD8+T-cell lymphoma. (A) Typical clinical presentation, with slowly progressive skin tumor on the right ear. (B) Diffuse proliferation of medium-sized pleomorphic cells in the dermis; the atypical cells strongly express CD8 (C) and TIA-1 (D). (E) CD68 shows a positive Golgi dot-like staining. Original magnification ×20 (B,E) and ×40 (C-D); hematoxylin and eosin (B) and immunoperoxidase (C-E) stain.
Primary cutaneous acral CD8+T-cell lymphoma. (A) Typical clinical presentation, with slowly progressive skin tumor on the right ear. (B) Diffuse proliferation of medium-sized pleomorphic cells in the dermis; the atypical cells strongly express CD8 (C) and TIA-1 (D). (E) CD68 shows a positive Golgi dot-like staining. Original magnification ×20 (B,E) and ×40 (C-D); hematoxylin and eosin (B) and immunoperoxidase (C-E) stain.
The prognosis of this condition is excellent; in typical cases, staging is not recommended.46,47 Skin lesions can easily be treated with surgical excision or radiotherapy. Cutaneous relapses may occur, but dissemination to extracutaneous sites is exceptional.50 Whether this condition should be labeled LPD or lymphoma is a matter of debate, but the majority of the participants at the Clinical Advisory Committee meeting favored the term “lymphoma.” However, recognition that these lesions have an indolent clinical behavior, despite an aggressive histology, is important to prevent unnecessarily aggressive treatment. Clinicopathologic correlation is essential to differentiate these cases from other types of CD8+ CTCL, such as primary cutaneous aggressive epidermotropic CD8+ T-cell lymphoma and cases of CD8+ MF.
Primary cutaneous CD4+ small/medium T-cell LPD
In the 2005 WHO-EORTC classification, primary cutaneous CD4+ small/medium T-cell lymphoma was included as a provisional type of CTCL, defined by a predominance of small- to medium-sized CD4+ pleomorphic T cells without prior or concurrent patches and plaques typical of MF.1 Patients typically present with a solitary plaque or tumor, generally on the face, neck, or upper trunk. Histologically, these lesions show dense, nodular to diffuse dermal infiltrates that consist mainly of CD4+ small-/medium-sized pleomorphic T cells, whereas a small proportion (<30%) of large pleomorphic cells may be present. These cells consistently express the follicular helper T-cell markers PD-1 (CD279), BCL6, and CXCL13 (Figure 4).51,52 The proliferation rate is low, varying between <5% and at most 20%. In almost all cases, there is a considerable admixture with small reactive CD8+ T cells, B cells, and histiocytes, including multinucleated giant cells. In some cases, monotypic plasma cells may be present.53 Patients have an excellent prognosis and, in typical cases, staging is not recommended. If skin lesions do not resolve spontaneously after biopsy, they should be treated primarily with intralesional steroids, surgical excision, or, in rare instances, with radiotherapy.52
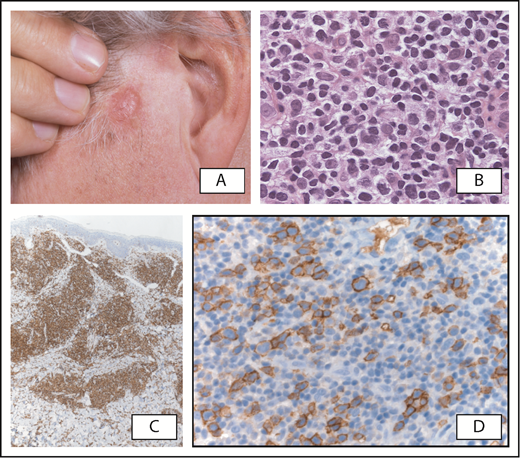
Primary cutaneous CD4+small/medium T-cell lymphoproliferative disorder. (A) Patient presenting with a tumor on the left cheek. (B) Detail of atypical dermal infiltrate showing a predominance of small/medium lymphoid cells and scattered large lymphoid cells, which express CD4 (C). (D) Expression of CD279/PD-1 by medium-sized to large atypical T cells, partly arranged in clusters. Original magnification ×200 (B,D) and ×40 (C); hematoxylin and eosin (B) and immunoperoxidase (C-D) stain.
Primary cutaneous CD4+small/medium T-cell lymphoproliferative disorder. (A) Patient presenting with a tumor on the left cheek. (B) Detail of atypical dermal infiltrate showing a predominance of small/medium lymphoid cells and scattered large lymphoid cells, which express CD4 (C). (D) Expression of CD279/PD-1 by medium-sized to large atypical T cells, partly arranged in clusters. Original magnification ×200 (B,D) and ×40 (C); hematoxylin and eosin (B) and immunoperoxidase (C-D) stain.
These cases have the same clinicopathologic and immunophenotypic features and the same clinical presentation and benign clinical course as cases previously referred to as nodular cutaneous pseudo-T-cell lymphomas; it is highly uncertain if they represent a frank malignancy.52,54 In the 2016 revision of the WHO classification and in the updated WHO-EORTC classification, the term “primary cutaneous CD4+ small/medium T-cell lymphoproliferative disorder” rather than lymphoma is therefore preferred.
Rare cases presenting with generalized skin lesions and large, rapidly growing tumors showing >30% large pleomorphic T cells and/or a high proliferative fraction on histopathology do not belong to this category.55,56 Such cases usually have a more aggressive clinical behavior and should be classified as peripheral T-cell lymphoma, NOS.
CBCL
In the 2005 WHO-EORTC classification, 3 main types of CBCL are recognized: primary cutaneous marginal zone lymphoma (PCMZL), primary cutaneous follicle center lymphoma (PCFCL), and primary cutaneous large B-cell lymphoma, leg type (PCDLBCL, LT).1 PCFCL and PCDLBCL, LT, were included as separate entities in the 2008 WHO classification for Tumors of Hematopoietic and Lymphoid Tissues and in its 2016 revision.3 In contrast, PCMZL was not categorized separately, but was included in the broad group of extranodal marginal zone lymphomas of mucosa-associated lymphoid tissue (MALT lymphoma), notwithstanding differences in histology, genetic profile, and clinical behavior. EBV+ mucocutaneous ulcer has been included as a new provisional entity in the 2016 revision of the WHO classification and the updated WHO-EORTC classification.
PCMZL
PCMZL particularly affects young adults and presents with solitary or, more commonly, multifocal plaques or nodules localized preferentially on the trunk and arms. Cutaneous relapses are common, in particular in patients presenting with multifocal skin lesions, but dissemination to extracutaneous sites is rarely observed. PCMZL have a very indolent clinical course and an excellent prognosis with a 5-year disease-specific survival rate close to 100%.57,58
Recent studies suggest the existence of 2 types of PCMZL.59,60 Unlike most other MALT lymphomas, the vast majority of PCMZLs express class-switched immunoglobulins, including immunoglobulin G (IgG), IgA, and IgE, and do not express the chemokine receptor CXCR3, which has been suggested to play a role in the homing of the malignant B cells to mucosa-associated malignant tissue. These cases show a predominance of T cells and only a small proportion of neoplastic B cells. Monotypic plasma cells are usually located at the periphery of the infiltrates and in the superficial dermis beneath the epidermis. Unlike MZL occurring at other sites, these class-switched cases do not show colonization of reactive germinal centers by neoplastic B cells, lymphoepithelial lesions, or transformation into a diffuse large B-cell lymphoma. A small subset of PCMZL shows a diffuse proliferation or large nodules of neoplastic B cells that express IgM and often CXCR3. These cases share many features with MALT lymphomas at other extranodal sites and are more likely to have extracutaneous disease. The class-switched cases are regarded by some authors as a clonal chronic cutaneous LPD rather than an overt lymphoma.61 This is further supported by clinical and histological similarities between PCMZL and pseudo-B-cell lymphoma (cutaneous lymphoid hyperplasia). The observation that both conditions may develop from chronic stimulation by intradermally applied antigens, such as tattoo pigments, tick bites, or antigen injections, suggests that they represent a continuous spectrum of cutaneous B-cell proliferations.62,63
PCFCL; PCDLBCL, LT; and PCDLBCL, other
There has always been much discussion regarding the classification of CBCLs with histologic features of a DLBCL. In the 2005 WHO-EORTC classification, 2 types were recognized: PCDLBCL, LT, and PCFCL with a diffuse growth pattern. In addition, there was a category PCDLBCL, other. Differentiation between PCDLBCL, LT, and PCFCL is extremely important because they have a different prognosis and require a different therapeutic approach. PCFCL is a tumor of neoplastic follicle center cells, often with a predominance of large centrocytes that generally present with localized skin lesions on the head or trunk, and can easily be managed by local radiotherapy and have an excellent prognosis. PCDLBCL, LT, is a more aggressive type of CBCL, histologically characterized by a monotonous proliferation of centroblasts and/or immunoblasts. These lymphomas particularly affect elderly women and present with generally rapidly growing tumors on 1 or both (lower) legs, or in ∼15% to 20% at sites other than the legs. Compared with PCFCL, they more often disseminate to extracutaneous sites and have a more unfavorable prognosis.57,64 In addition to clinical and histological criteria, differences in immunophenotype and genetic aberrations may be helpful in distinguishing both conditions (Table 4). In contrast to PCFCL, PCDLBCL, LT, strongly expresses BCL2, IRF4/MUM1, and IgM; recent studies reported expression of MYC in 65%.65-68 Because >90% of PCDLBCL, LT, cases express BCL2, double expression of MYC and BCL2 is also present in two-thirds of PCDLBCL, LT, cases. Detection of double expression may facilitate differentiation from PCFCL (Figure 5).68 Rearrangements of the MYC gene have been detected in 30% of PCDLBCL, LT, cases, with a second rearrangement in the BCL6 gene in rare cases.68
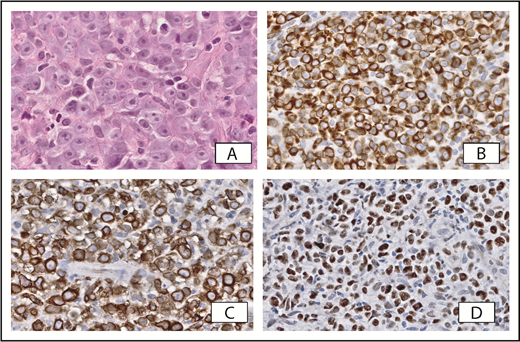
Primary cutaneous diffuse large B-cell lymphoma, leg type. (A) Cohesive sheets of large transformed cells with prominent nucleoli. Strong expression of BCL2 (B), IgM (C), and MYC (D) may facilitate differentiation from PCFCL. Original magnification ×400 (A, hematoxylin and eosin stain) and ×200 (B-D, immunoperoxidase stain).
Primary cutaneous diffuse large B-cell lymphoma, leg type. (A) Cohesive sheets of large transformed cells with prominent nucleoli. Strong expression of BCL2 (B), IgM (C), and MYC (D) may facilitate differentiation from PCFCL. Original magnification ×400 (A, hematoxylin and eosin stain) and ×200 (B-D, immunoperoxidase stain).
In the past decade, the results of genetic studies have contributed to a better understanding of the molecular mechanisms involved in the pathogenesis of these lymphomas and provided additional diagnostic and prognostic markers. Loss of CDKN2A either by gene deletion or promoter methylation and the presence of MYD88 L265P mutations, both observed in about two-thirds of patients with PCDLBCL, LT, have been reported to be associated with an inferior prognosis.69,70 The presence of MYD88 L265P mutations and mutations in different components of the B-cell receptor signaling pathway, including CARD11 (10%), CD79B (20%), and TNFAIP3/A20 (40%), strongly suggest constitutive NF-κB activation in PCDLBCL, LT.71,72 The mutational profile of PCDLBCL, LT, including NF-κB–activating mutations and PDL1/PDL2 translocations, overlaps with that of the ABC subtype of systemic DLBCL, NOS, but is most similar to that of primary central nervous system lymphomas and primary testicular lymphomas.73 In contrast to PCDLBCL, LT, MYD88 L265P mutation is absent and loss or inactivation of CDKN2A is not or is rarely found in PCFCL.74,75 PCDLBCL, LT, should also be distinguished from iatrogenic immunodeficiency–associated LPD (see the following section) and secondary cutaneous DLBCL. In all cases, adequate staging is therefore required.
The term PCDLBCL, other, was introduced in the 2005 WHO-EORTC classification as an encompassing term for rare cases of DLBCL first presenting in the skin that could not be classified as either PCDLBCL, LT, or PCFCL. This term, however, has been interpreted and used in different ways and has been the source of much confusion. It has been used for cases composed of large transformed cells that, unlike PCDLBCL, LT, were negative for BCL2, or for cases of PCDLBCL that could not be classified properly using the Hans algorithm.65,67 However, there are no significant differences between PCDLBCL, LT, with or rare cases without expression of BCL2, and categorization of BCL2− cases as PCDLBCL, other, is therefore not justified.57,65,76 Moreover, PCDLBCL, LT, and PCFCL were already defined on the basis of a combination of clinical, histological, immunophenotypical, and genetic criteria long before the Hans algorithm was developed. To avoid further confusion, the 2018 update of the WHO-EORTC classification, as with the prior WHO classifications, does not contain a separate category of PCDLBCL, other, anymore. In rare cases that cannot be classified as either PCDLBCL, LT, or PCFCL, a diagnosis of primary cutaneous DLBCL, NOS, should be made.
Intravascular large B-cell lymphoma is a rare disease defined by an accumulation of large neoplastic B cells within the lumina of blood vessels. These lymphomas typically affect the central nervous system, lungs, and skin, and are generally associated with a poor prognosis. A cutaneous variant presenting with skin-limited disease at the time of diagnosis has been described. It accounts for ∼25% of all cases in the Western world, predominantly affects females, and has a much better prognosis than for patients with systemic disease.
EBVMCU and other cutaneous immunodeficiency–associated B-cell LPDs
EBV+ mucocutaneous ulcer (EBVMCU) is defined as a solitary, sharply demarcated ulcerating lesion involving the skin, oropharyngeal mucosa, or gastrointestinal tract in patients with age-related or iatrogenic immunosuppression (methotrexate, azathioprine, cyclosporine, tumor necrosis factor inhibitors). Histologically, the lesions contain large Hodgkin-like EBV+ B cells in a mixed inflammatory background. These large transformed cells are PAX5+, show variable expression of CD20, display a nongerminal center phenotype (IRF4/MUM1+, CD10−, BCL6−) and typically express CD30 with coexpression of CD15 in almost one-half of the cases. EBVMCU usually runs a self-limited, indolent course. In iatrogenic cases, reduction of immunosuppressive therapy without additional chemotherapy or radiotherapy may result in complete remission.77
Apart from EBVMCU, there are other cutaneous manifestations of B-cell LPDs occurring in the setting of iatrogenic immunosuppression: skin lesions may be EBV+ or EBV− and both can be solitary or generalized with or without ulceration.78 Methotrexate-associated B-cell LPDs usually show the histology of a DLBCL or Hodgkin-like features.3 The latter is particularly seen in EBV+ cases, which account for 40% to 50% of all cases.79 Recognition of these iatrogenic immunodeficiency–associated lesions is important, because in all cases reduction or cessation of immunosuppressive treatment may result in (complete) remissions and should be attempted before more aggressive therapy is considered.78-80
In conclusion, since the publication of the first WHO-EORTC classification, much progress has been made, and this 2018 update continues to be a useful guide for clinicians involved in the care of patients with a cutaneous lymphoma. Genome-wide genetic studies have contributed to a better understanding of the molecular pathways involved in the pathogenesis of the different types of cutaneous lymphomas and resulted in the recognition of additional diagnostic and prognostic criteria and new potential therapeutic targets. Although genetic markers may become increasingly important, integration of histologic, immunophenotypic, genetic, and, in particular in case of cutaneous lymphomas, clinical data remain essential for an accurate diagnosis. In past decades, a multidisciplinary approach with collaboration among pathologists, dermatologists, hematologists, and radiation oncologists has been crucial for defining new entities and classifications, and is the best guarantee for further progress in the diagnosis and treatment of patients with a cutaneous lymphoma.
Authorship
Contribution: All authors contributed to the contents of this manuscript, which was written by R.W., W.K., E.B., F.F., and E.S.J. and reviewed and edited by all authors.
Conflict-of-interest disclosure: R.W. is a member of the Scientific Advisory Board of Takeda. The remaining authors declare no competing financial interests.
Correspondence: Rein Willemze, Department of Dermatology; B1-Q90, Leiden University Medical Center, PO Box 9600, 2300 RC Leiden, The Netherlands; e-mail: rein.willemze@planet.nl.