Key Points
CD37 CAR T cells undergo activation, proliferation, and cytokine production in response to CD37 engagement alone, and in bispecific format.
Anti-CD37 CAR T cells have antitumor activity against B- and T-cell lymphomas without fratricide.

Abstract
Chimeric antigen receptor (CAR) T cells have emerged as a novel form of treatment of patients with B-cell malignancies. In particular, anti-CD19 CAR T-cell therapy has effected impressive clinical responses in B-cell acute lymphoblastic leukemia and diffuse large B-cell lymphoma. However, not all patients respond, and relapse with antigen loss has been observed in all patient subsets. Here, we report on the design and optimization of a novel CAR directed to the surface antigen CD37, which is expressed in B-cell non-Hodgkin lymphomas, in chronic lymphocytic leukemia, and in some cases of cutaneous and peripheral T-cell lymphomas. We found that CAR-37 T cells demonstrated antigen-specific activation, cytokine production, and cytotoxic activity in models of B- and T-cell lymphomas in vitro and in vivo, including patient-derived xenografts. Taken together, these results are the first showing that T cells expressing anti-CD37 CAR have substantial activity against 2 different lymphoid lineages, without evidence of significant T-cell fratricide. Furthermore, anti-CD37 CARs were readily combined with anti-CD19 CARs to generate dual-specific CAR T cells capable of recognizing CD19 and CD37 alone or in combination. Our findings indicate that CD37-CAR T cells represent a novel therapeutic agent for the treatment of patients with CD37-expressing lymphoid malignancies.
Introduction
Non-Hodgkin lymphoma (NHL) is a heterogeneous group of malignancies including B-l and T-cell lymphomas, accounting for about 4% of all tumors.1 Approximately 80% of NHLs are derived from the B-cell lineage and uniformly express B-cell differentiation antigens, including CD19 and CD20. These surface antigens represent pivotal targets for antibody-based therapeutics and for chimeric antigen receptor (CAR) T-cell therapies. Anti-CD19 CAR T-cell treatment has effected responses in the 60% to 80% range, and approximately 40% of patients have achieved long-term complete remissions.2-8 As of this writing, 2 anti-CD19 CAR T-cell products, axicabtagene ciloleucel, which bears the CD28 costimulatory domain, and tisagenlecleucel, in which the CAR includes the 4-1BB costimulatory domain, have been approved for the treatment of relapsed or refractory large-cell lymphomas. The tisagenlecleucel CAR T-cell product has also been approved for the treatment of relapsed or refractory acute B-cell lymphoblastic leukemia in children and young adults. However, disease relapse resulting from CD19 antigen target loss has been observed in both patients with acute lymphoblastic leukemia (ALL) and patients with NHL,2,9,10 and represents a new unmet clinical need. Thus, even in B-cell lymphomas, there is a need to target alternative surface antigens with CAR T cells.11
CD37 is a 4-passage transmembrane protein of the tetraspanin superfamily. Although its biologic function is incompletely understood, CD37 is involved in various different cellular processes, including survival, proliferation, adhesion, and migration of lymphocytes.12-16 CD37 expression is restricted to lymphoid tissues, and in particular to mature B cells, with low levels of expression on plasma cells and dendritic cells.17,18 This pattern is mirrored in B-cell malignancies: it is expressed in mature B-cell neoplasms, including mantle cell lymphoma (MCL), follicular lymphoma, diffuse large B-cell lymphoma (DLBCL), Burkitt’s lymphoma, and chronic lymphocytic leukemia (CLL), whereas it is low or absent in ALL and multiple myeloma.17 Interestingly, recent studies have reported CD37 expression in cutaneous and peripheral T-cell lymphoma samples (CTCL and PTCL).19 These patients have a poor prognosis and are underserved by current therapies, making this a high-priority set of diseases for the development of CAR T-cell approaches.20-22 CD37 represents a promising target for B- and T-cell lymphoma therapy, and recently has been validated as a druggable target, using monoclonal antibodies and antibody-drug conjugates in clinical trials of both B- and T-cell lymphoma.19,23,24
Here, we confirmed expression of CD37 in B- and T-cell malignancies, generated a novel CAR targeting CD37, characterized its activity in a range of cells with varying levels of antigen density, and used a series of preclinical models to assess its efficacy. We demonstrate that CAR-37 engenders antitumor effect in vitro and leads to prolonged remissions in cell line-based and patient-derived xenograft (PDX) models of NHL. CAR-37 T cells were also active against T-cell lymphomas. Despite reports of broader expression of CD37 on other immune cells, we did not observe CAR-37 T-cell activation or degranulation in response to coculture with other immune cells. We found that CD37 CAR T cells appear to be as effective as CD19-targeted CAR T cells in B-cell lymphomas in vitro and in xenograft models. Finally, combinations of CD37 and CD19 in a bispecific CAR format are readily activated and effective in vitro and in vivo, thus enabling the use of dual-targeting CAR T cells for lymphoma.
Materials and methods
Construction of CARs and T-cell culture transduction
Two anti-CD37 CAR constructs were synthesized and cloned into a third-generation lentiviral plasmid backbone under the regulation of a human EF-1α promoter. All the CARs bear a CD8 hinge, 4-1BB costimulatory domain, and CD3 ζ signaling domain. Vectors also contained a second transgene coding for the fluorescent reporter mCherry to facilitate enumeration of transduction efficiency. Human T cells were purified (Stem Cell Technologies, Catalog #15061) from anonymous human healthy donor leukopacs purchased from the Massachusetts General Hospital blood bank under an Institutional Review Board–exempt protocol. Details of primary T-cell culture are provided in the supplemental Methods, available on the Blood Web site.
Cellular cytotoxicity and cytokine assays
For cytotoxicity assays, CAR T-cell effector cells were cocultured with click beetle green (CBG) luciferase-expressing tumor targets at the indicated ratios for 16 hours. Luciferase activity was measured with a Synergy Neo2 luminescence microplate reader (Biotek). For the analysis of soluble cytokines, effector cells were cocultured with tumor targets at a 1:1 ratio for 24 hours. Additional information is provided in supplemental Methods.
In vivo studies
All animal procedures were performed in accordance with Federal and Institutional Animal Care and Use Committee requirements under a protocol approved at Massachusetts General Hospital. Detailed methods are provided in supplemental Methods.
Further methods are available in supplemental Data.
Results
CD37 is highly expressed on human lymphoma
We used flow cytometry to examine CD37 and CD19 expression on the leukemia and NHL cell lines Nalm6, Jeko-1, and Raji (Figure 1A), and in patient-derived MCL lines (Figure 1B-C) and primary patient CLL cells (Figure 1D). We also generated K562 cells transduced with both CD37 and CD19 to use as artificial antigen presenting cells for in vitro stimulation and a positive control for cytotoxicity assays (Figure 1A). Although pre-B-cell-derived leukemia cells (Nalm6) expressed CD19 but not CD37 (Figure 1A), all the lymphoma cells expressed both CD19 and CD37 (Figure 1A-C). In the patient-derived MCL samples, we noted high and uniform expression of CD37, even higher than CD19, based on mean fluorescence intensity of the gated positive cells (Figure 1C), although we recognize that this difference could reflect antibody binding differences or brightness of the fluorophores.

CD37 is highly expressed on lymphoma cell lines and patient-derived samples. (A) Fluorescence-activated cell sorter (FACS) plots of tumor cell lines stained with CD37 and CD19 antibodies. NALM6, acute lymphoblastic leukemia cell line; K562 expressing CD19 and CD37, positive control; JEKO-1, MCL cell line; RAJI, Burkitt lymphoma cell line. (B) FACS plots of samples derived from 3 patients with MCL. (C) MFI of CD19 and CD37 on MCL patient’s cells. Each dot represents a separate xenograft sample (n = 3; medians shown). (D) CD19 and CD37 expression on PBMC from patients with CLL gated on CD3− lymphocytes (n = 20; mean ± SD shown; ****P < .0001 by Student t test). (E) Distribution of CD19 and CD37 antigens on CLL PBMC cells. Antibodies bound per cell gated on CD3− cells from each patient sample is shown. (F) Median of CD19 and CD37 antigen density is shown. CD19 range, 24 396 to 70 952; mean, 43 238; CD37 range, 8992 to 46 550; mean, 23 989. (G) CD37 immunohistochemistry in primary ALK-negative (left) and ALK-positive (right) ALCL specimens from the tissue microarray. Original magnification ×100.
CD37 is highly expressed on lymphoma cell lines and patient-derived samples. (A) Fluorescence-activated cell sorter (FACS) plots of tumor cell lines stained with CD37 and CD19 antibodies. NALM6, acute lymphoblastic leukemia cell line; K562 expressing CD19 and CD37, positive control; JEKO-1, MCL cell line; RAJI, Burkitt lymphoma cell line. (B) FACS plots of samples derived from 3 patients with MCL. (C) MFI of CD19 and CD37 on MCL patient’s cells. Each dot represents a separate xenograft sample (n = 3; medians shown). (D) CD19 and CD37 expression on PBMC from patients with CLL gated on CD3− lymphocytes (n = 20; mean ± SD shown; ****P < .0001 by Student t test). (E) Distribution of CD19 and CD37 antigens on CLL PBMC cells. Antibodies bound per cell gated on CD3− cells from each patient sample is shown. (F) Median of CD19 and CD37 antigen density is shown. CD19 range, 24 396 to 70 952; mean, 43 238; CD37 range, 8992 to 46 550; mean, 23 989. (G) CD37 immunohistochemistry in primary ALK-negative (left) and ALK-positive (right) ALCL specimens from the tissue microarray. Original magnification ×100.
We next evaluated CD37 and CD19 expression on PBMC derived from 21 patients with chronic lymphocytic leukemia by flow cytometry (Figure 1D), and again noted higher and more uniform expression of CD37 among the CD3-negative lymphocytes compared with CD19. When gated on the CD3−CD20+ B cells, CD37 expression remained high and uniform (supplemental Figure 1). To determine the level of antigen density for both antigens, we quantified the expression of CD19 and CD37 on these 21 samples, using beads (Figure 1E). We found that antigen density on a per cell basis was higher for CD19 than for CD37 (mean = 31 829 ± 3212 antibodies bound per cell for CD19 vs 29 680 ± 3232 antibodies bound per cell for CD37; Figure 1F; supplemental Table 1). Based on 29 samples of bone marrow aspirates, lymph node biopsies, and peripheral blood from patients with hematologic malignancies, we confirmed expression of CD37 on B-cell lymphomas and normal B cells, and not on hematopoietic stem cells from normal donors or hematogones (supplemental Table 2).
In this data set, there were no samples from patients with T-cell malignancies. Thus, we assessed expression of CD37 in primary peripheral T-cell lymphomas by performing immunohistochemical staining on a tissue microarray containing triplicate cores of 67 PTCL samples from 9 different subtypes. Overall, positive staining in at least a subset of cells was seen in patient samples from each subtype, including 15 of 16 angioimmunoblastic T-cell lymphomas; 1 of 1 ALK+ anaplastic large cell lymphomas (ALCLs); 6 of 13 ALK-negative ALCLs; 2 of 6 adult T-cell leukemias; 18 of 23 PTCL, not otherwise specified; 1 of 1 enteropathy-associated T-cell lymphoma; 4 of 4 extranodal NK/T-cell lymphoma, nasal type; 1 of 1 T-cell prolymphocytic leukemia; and 1 of 2 hepatosplenic γ/δ T-cell lymphomas. Representative strong staining for CD37 in a PTCL (ALCL) sample is shown in Figure 1G.
Generation of anti-CD37 CAR T cells
We designed 2 anti-CD37 chimeric antigen receptors consisting of anti-CD37 scFv and CD8 transmembrane domain in tandem with 41BB intracellular signaling domain and CD3 ζ (Figure 2A). The scFv’s were synthesized in both orientations of the variable heavy and light chains, generating CAR-37 L-H and CAR-37 H-L. To facilitate evaluation of transduction efficiency, we incorporated the mCherry fluorescent reporter gene after a 2A ribosomal skip sequence at the C-terminal of the CAR sequence. High-efficiency gene transfer into primary activated human T cells was obtained with both constructs using third-generation self-inactivating lentiviral vectors (Figure 2B-C). CAR-37 T cells displayed expansion after initial priming with anti-CD3/CD28 beads during the first 10 days, comparable to anti-CD19 CAR T cells (CAR-19). As a comparator, we generated CAR-19 T cells based on the same backbone, with the CD8 transmembrane domain and 4-1BB and CD3zeta intracellular signaling domains. We found that CAR-37 T cells could undergo long-term expansion through repetitive antigen stimulations with irradiated K562 cells transduced to express CD37 and CD19 (Figure 2D). We then tested the activation of our CARs, using Jurkat reporter (NFAT-Luciferase) T-cells. After transducing the Jurkat reporter cells with the different CAR constructs, we cocultured them with a variety of stimuli, including anti-CD3/CD28 beads, B-cell lymphoma tumor cells, Nalm6 leukemia cells, or media as a negative control. Measurements of the luminescence demonstrated specific T-cell activation and NFAT-mediated luminescence in response to antigen stimulation (Figure 2E). In this assay, anti-CD37 CARs in the L-H orientation appeared to initiate activation more robustly than anti-CD37 CARs in the H-L orientation, and at similar levels as anti-CD19 CARs in response to CD19-expressing tumors. However, there is no known threshold or optimal amount of NFAT translocation that corresponds to an optimal CAR construct, and we interpret these findings with caution. These data indicate that anti-CD37 CARs mediate T-cell activation signals in response to specific antigen stimulation and can undergo long-term growth in response to antigen stimulation.

In vitro CAR-37 generation and expansion. (A) Two anti-CD37 second-generation chimeric antigen receptors were constructed with different orientations of a humanized murine antibody-derived single-chain variable fragment: the light-to-heavy orientation (CAR-37 L-H, top) and the heavy-to-light (CAR-37 H-L, bottom). (B) Representative flow plots of primary human T cells transduction efficiency after 10 days of activation with CD3/CD28 beads. (C) Expanded T cells from 3 healthy donors included variable CAR-37 expression with a mean of 38% (L-H) and 75% (H-L). (D) Ex vivo expansion of CD3/CD28 bead-activated and target-stimulated T-cells using static culture conditions in 3 healthy donors for 38 days. Each arrow represents antigen stimulation with K562 cells transduced to express CD37 and CD19. (E) Activation of Jurkat reporter (NFAT-Luc) T cells transduced with different CAR constructs and cocultured with tumor cells. Luciferase activity was measured after 16 hours. (CD3-CD28 beads: positive control). (F) Number of carboxyfluorescein succinimidyl ester–labeled, unstimulated target T cells measured by flow cytometry after 24 hours of coculture at indicated E:T ratio with CAR-37H-L, CAR-19, or UTD T cells. (G) Number of carboxyfluorescein succinimidyl ester–labeled T cells stimulated with PMA/ionomycin for 6 hours, then measured by flow cytometry after 24 hours of coculture at indicated E:T ratio with CAR-37H-L, CAR-19, or UTD T cells. (H) Number of Jeko-1 CBG−GFP cells measured by flow cytometry after 24 hours of coculture at indicated E:T ratio with CAR-37H-L, CAR-19 or untransduced T cells. Bars indicate mean ± standard error of the mean (SEM) count of triplicates from 1 normal donor, representative of 3 normal donors. (I) CD107a and IFN-γ production relative to media by CAR-37H-L, CAR-19 T cells incubated with primary immune cells for 6 hours at 1:1 E:T ratio was analyzed by flow cytometry. Bars show mean ± SEM percentage of the 3 normal donors analyzed.
In vitro CAR-37 generation and expansion. (A) Two anti-CD37 second-generation chimeric antigen receptors were constructed with different orientations of a humanized murine antibody-derived single-chain variable fragment: the light-to-heavy orientation (CAR-37 L-H, top) and the heavy-to-light (CAR-37 H-L, bottom). (B) Representative flow plots of primary human T cells transduction efficiency after 10 days of activation with CD3/CD28 beads. (C) Expanded T cells from 3 healthy donors included variable CAR-37 expression with a mean of 38% (L-H) and 75% (H-L). (D) Ex vivo expansion of CD3/CD28 bead-activated and target-stimulated T-cells using static culture conditions in 3 healthy donors for 38 days. Each arrow represents antigen stimulation with K562 cells transduced to express CD37 and CD19. (E) Activation of Jurkat reporter (NFAT-Luc) T cells transduced with different CAR constructs and cocultured with tumor cells. Luciferase activity was measured after 16 hours. (CD3-CD28 beads: positive control). (F) Number of carboxyfluorescein succinimidyl ester–labeled, unstimulated target T cells measured by flow cytometry after 24 hours of coculture at indicated E:T ratio with CAR-37H-L, CAR-19, or UTD T cells. (G) Number of carboxyfluorescein succinimidyl ester–labeled T cells stimulated with PMA/ionomycin for 6 hours, then measured by flow cytometry after 24 hours of coculture at indicated E:T ratio with CAR-37H-L, CAR-19, or UTD T cells. (H) Number of Jeko-1 CBG−GFP cells measured by flow cytometry after 24 hours of coculture at indicated E:T ratio with CAR-37H-L, CAR-19 or untransduced T cells. Bars indicate mean ± standard error of the mean (SEM) count of triplicates from 1 normal donor, representative of 3 normal donors. (I) CD107a and IFN-γ production relative to media by CAR-37H-L, CAR-19 T cells incubated with primary immune cells for 6 hours at 1:1 E:T ratio was analyzed by flow cytometry. Bars show mean ± SEM percentage of the 3 normal donors analyzed.
Because CD37 has been reportedly expressed on T cells and other hematopoietic mononuclear cells, we interrogated expression of CD37 on whole-blood immune cells from healthy donors (supplemental Figure 2). We found high expression of CD37 on B cells, with minimal expression on monocytes, but not NK cells or T cells. However, many T-cell markers change with activation, and we tested for the possibility of fratricide by coculturing CAR-37 T cells with activated or nonactivated carboxyfluorescein diacetate succinimidyl ester-labeled T cells from the same donors. After 24 hours, we analyzed target T-cell counts. We did not detect any significant difference in the counts of labeled resting T cells (Figure 2F) or labeled activated T cells (Figure 2G) that had been cocultured with CAR-37 T cells compared with those cultured with CAR-19 T cells, despite expected cytotoxicity against Jeko-1 target cells in the same experiment (Figure 2H). Next, we tested degranulation and interferon-γ (IFN-γ) production of CAR-37 T cells against monocytes, NK cells, and in vitro differentiated M1 or M2 macrophages (Figure 2I). We found no significant difference in degranulation or IFN-γ production between CAR-37 and CAR-19 T cells, indicating no evidence of immune cell toxicity induced by CAR-37 T cells.
CAR-37 T cells exhibit robust effector functions in response to CD37-positive tumor cells in vitro
To define the antitumor activity of CAR-37 T cells, we first performed cytotoxicity assays against a panel of lymphoma cell lines. CAR-37 T cells were cocultured with Jeko-1, OSU-CLL, Raji, or K562-CD37-CD19 cells at various effector to target ratios for 16 hours (Figure 3A). All CAR-37 T-cell effector were able to lyse target cells, but in contrast to what we observed in the Jurkat activation assay, the heavy-light chain configuration was more favorable than the light-heavy configuration for anti-CD37 CAR T cells. This difference was evident and consistent with all the tumor lines tested. Notably, T cells transduced with CAR-37 H-L or CAR-19 demonstrated equivalent cytolytic activity against these target tumor cells, all of which express both antigens.
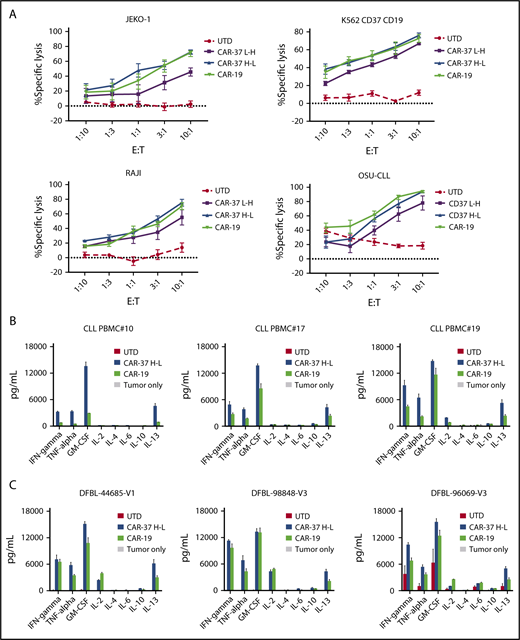
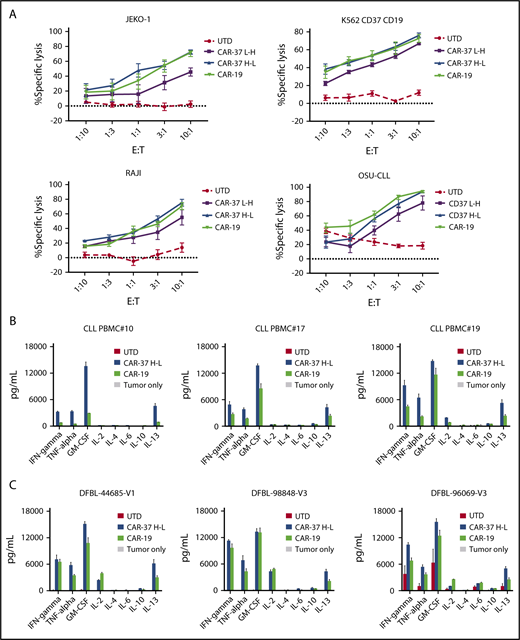
CAR-37 T cells exhibit robust in vitro effector functions in response to CD37 positive tumor cells. (A) Cytotoxic capacity of CAR-37 T cells was measured after overnight coculture with targets. CAR T cells were cocultured at indicated E:T ratios with indicated tumor cell lines. Increasing concentration of CAR-37 and CAR-19 T cells led to specific killing, whereas no killing was observed in the control group (UTD). The cytotoxicity assay is representative of 3 independent experiments conducted with different healthy donors. Cytokine production by CAR-37H-L, CAR-19, or UTD T cells incubated with primary CLL (B) or MCL PDX (C) tumor samples. CAR T cells were incubated with target cells for 24 hours at a 1:1 E:T ratio, and culture supernatants were analyzed by Luminex assay. Data are plotted as mean ± SEM for 3 donors.
CAR-37 T cells exhibit robust in vitro effector functions in response to CD37 positive tumor cells. (A) Cytotoxic capacity of CAR-37 T cells was measured after overnight coculture with targets. CAR T cells were cocultured at indicated E:T ratios with indicated tumor cell lines. Increasing concentration of CAR-37 and CAR-19 T cells led to specific killing, whereas no killing was observed in the control group (UTD). The cytotoxicity assay is representative of 3 independent experiments conducted with different healthy donors. Cytokine production by CAR-37H-L, CAR-19, or UTD T cells incubated with primary CLL (B) or MCL PDX (C) tumor samples. CAR T cells were incubated with target cells for 24 hours at a 1:1 E:T ratio, and culture supernatants were analyzed by Luminex assay. Data are plotted as mean ± SEM for 3 donors.
Next, we analyzed cytokine production in response to antigen stimulation. We compared the pattern of cytokines produced by different CAR constructs after stimulation with target cells. CAR-37 T cells demonstrated antigen-specific production of the Th1-type cytokines tumor necrosis factor-α, IFN-γ, interleukin 2, and granulocyte-macrophage colony-stimulating factor after in vitro stimulation with tumor cell lines (supplemental Figure 3A), primary CLL (Figure 3B), and MCL PDX samples (Figure 3C). Consistent with the cytotoxicity assays, these experiments demonstrate improved antigen-specific effector function of CAR-37 H-L compared with CAR-37 L-H.
CAR-37 T cells eradicate MCL tumor in vivo
Because our in vitro assays indicated that CAR-37 H-L was possibly but not definitively superior to CAR-37 L-H, we compared these 2 formats in a xenogeneic model of MCL. Nonobese scid common gamma chain knockout (NSG) mice were injected intravenously with luciferase-expressing Jeko-1 (CBG−GFP+) cells. Seven days later, disease burden was assessed by BLI in all mice, and CAR-37 or untransduced (UTD) T cells were administered by tail vein injection. By 14 days, there was partial disease control in CAR-37 L-H treated animals, but complete disease eradication in CAR-37 H-L treated mice (supplemental Figure 4), thus confirming the superior antigen-induced effector function of CAR37 H-L over CAR37 L-H, which we then selected for further experiments and for the direct comparison with CAR-19 T cells in vivo.
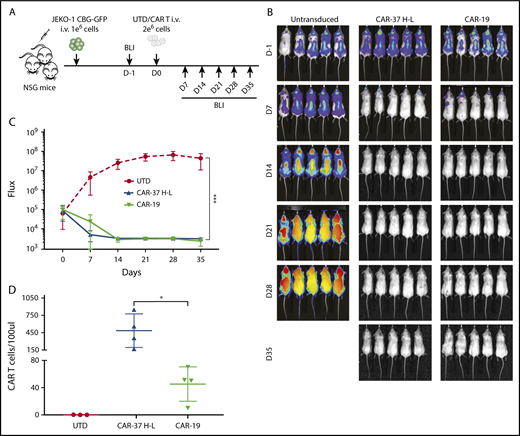
We injected CAR T cells into NSG mice 7 days after intravenous injection of the MCL cell line Jeko-1 (CBG−GFP+), as shown (Figure 4A). Serial imaging of luminescence to assess tumor burden indicated rapid and complete elimination of tumor by day 14 for both CAR-37 and CAR-19 T cells, with dramatic reductions in tumor volume by day 7 (Figure 4B-C). We confirmed CAR T-cell persistence in the peripheral blood by flow cytometry (Figure 4D), with greater persistence of CAR-37 T cells at day 7 (P < .05).
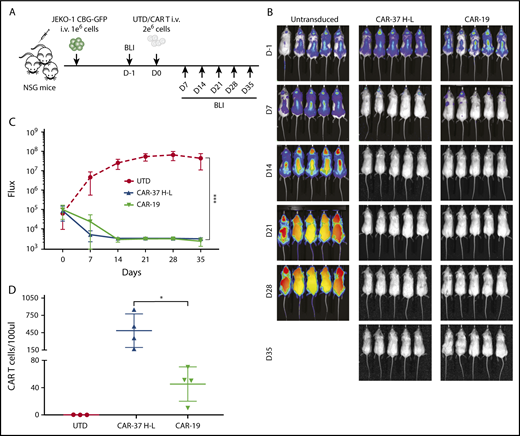
In vivo CAR-37 mediated tumor clearance of a MCL model. (A) Experiment schematic: NSG mice were injected IV with 1 × 106 JEKO-1(CBG−GFP) cells and monitored by BLI for tumor burden at different points. At day 0, mice were randomly assigned on the basis of tumor burden (BLI) to receive 2 × 106 control T cells (UTD), CAR-37, or CAR-19. (B) Representative bioluminescent images of JEKO-1 growth over time. (C) Average flux (photons/s) of whole mice in the 3 groups at different points. Graph is representative of 2 experiments with 5 mice per group, conducted with CAR T cells obtained from 2 different healthy donors. Mean ± SD shown. ***P < .001 by 2-way analysis of variance. (D) Absolute numbers of CAR T cells were monitored by bleeding and flow cytometric detection. Absolute CAR T-cell counts in peripheral blood at day 14 after CAR T injection are shown (Student t test, *P < .05).
In vivo CAR-37 mediated tumor clearance of a MCL model. (A) Experiment schematic: NSG mice were injected IV with 1 × 106 JEKO-1(CBG−GFP) cells and monitored by BLI for tumor burden at different points. At day 0, mice were randomly assigned on the basis of tumor burden (BLI) to receive 2 × 106 control T cells (UTD), CAR-37, or CAR-19. (B) Representative bioluminescent images of JEKO-1 growth over time. (C) Average flux (photons/s) of whole mice in the 3 groups at different points. Graph is representative of 2 experiments with 5 mice per group, conducted with CAR T cells obtained from 2 different healthy donors. Mean ± SD shown. ***P < .001 by 2-way analysis of variance. (D) Absolute numbers of CAR T cells were monitored by bleeding and flow cytometric detection. Absolute CAR T-cell counts in peripheral blood at day 14 after CAR T injection are shown (Student t test, *P < .05).
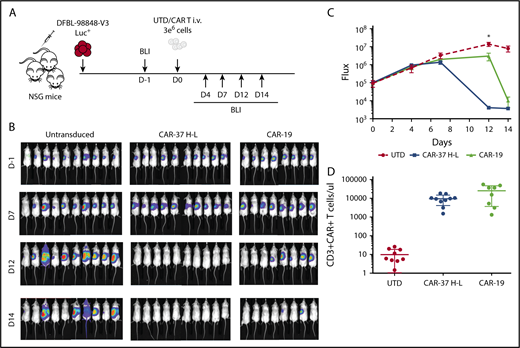
Although tumor cell lines are quite useful in the assessment of efficacy of CAR T cells, they often do not fully represent the heterogeneity and biology of primary patient tumors. In contrast, PDX models, in which tumor cells are derived directly from patients and cultured for only 2 to 3 passages, are thought to resemble the clinical setting more closely. We assessed the CAR-37 (H-L configuration) in PDX models of MCL. Ten NSG mice were injected with luciferase-expressing MCL-PDX cells. After confirming disease engraftment and tumor burden similar to the Jeko-1 model by BLI, we injected CAR-37, CAR-19, or UTD T cells (Figure 5A). We found that CAR-37 T cells were able to clear the tumor in only 12 days, notably faster than CAR-19 (P < .05 at the day 12 point; Figure 5B-C). Flow cytometry assessment of peripheral blood collected at day 14 confirmed the persistence of CAR T cells in the blood (Figure 5D). Collectively, these results indicate that CAR-37 T cells mediate significant antitumor effects against B-cell NHL in vivo, in both tumor-line and PDX-models of MCL.

CAR-37 mediated tumor clearance of MCL patient-derived xenograft. (A) Protocol schema: NSG mice were injected intravenously with 1 × 106 MCL patient-derived cells and monitored for tumor burden by bioluminescent imaging (BLI) over time. At day 0, mice were randomly assigned on the basis of tumor burden to receive 3 × 106 control T cells (UTD), CAR-37, or CAR-19. (B) Representative BLI of MCL xenografts over time. (C) Average flux (photons/s) of whole mice in the 3 groups at different points. Graph is representative 2 simultaneous experiments of 5 mice per group, conducted with CAR T cells obtained from 2 different healthy donors, and pooled data. Mean ± SD shown (Student t test, *P < .05). (D) Absolute numbers of CAR T cells were monitored in peripheral blood using flow cytometry. Absolute counts of CAR T cells are plotted at day 14.
CAR-37 mediated tumor clearance of MCL patient-derived xenograft. (A) Protocol schema: NSG mice were injected intravenously with 1 × 106 MCL patient-derived cells and monitored for tumor burden by bioluminescent imaging (BLI) over time. At day 0, mice were randomly assigned on the basis of tumor burden to receive 3 × 106 control T cells (UTD), CAR-37, or CAR-19. (B) Representative BLI of MCL xenografts over time. (C) Average flux (photons/s) of whole mice in the 3 groups at different points. Graph is representative 2 simultaneous experiments of 5 mice per group, conducted with CAR T cells obtained from 2 different healthy donors, and pooled data. Mean ± SD shown (Student t test, *P < .05). (D) Absolute numbers of CAR T cells were monitored in peripheral blood using flow cytometry. Absolute counts of CAR T cells are plotted at day 14.
Targeting CD37 on T-cell lymphoma
We analyzed the surface expression of CD37 in PTCL lines and PDX samples of PTCL by flow cytometry. We identified 3 cell lines (Hut78, Fedp, and Seax) and 5 PDX samples that expressed CD37 on the cell surface at varying levels (Figure 6A-B; supplemental Table 3). We tested the activation of CAR-37 T cells after coculture with these PTCL samples. The CD69 activation marker was analyzed by flow cytometry on gated CAR+ T cells; we noted robust upregulation of CD69 after 6 hours of coculture with PTCL target cells, indicating CAR37 T-cell activation (Figure 6C). Interestingly, the degree of CD69 upregulation was independent of the level of expression of the CD37 antigen on the target cells. We then tested the degranulation of CAR-37 T cells by measuring expression of CD107a in CAR-37 cells after incubation with PTCL target cells (Figure 6D). Consistent with the CD69 assays, we found that CAR-37 T cells degranulated in response to PTCL cells, indicating activation of CAR37 T cells in response to PTCL. Finally, CAR-37 T cells effectively lysed the CD37-positive PTCL cell lines Hut78 and Fepd, as determined by cytotoxicity assays performed at varying E:T ratios (Figure 6E-F). Taken together, these experiments demonstrate that CAR-37 T cells are activated against and lyse T-cell lymphoma cells.

CAR-37 in vitro activity against T-cell lymphoma and leukemia. (A) CD37 expression on PTCL tumor cell lines. (B) Representative FACS plots from patient-derived samples. (C) CD69 expression and (D) CD107a degranulation of CAR T cells, as evaluated by flow cytometry after 6 hours of coculture with indicated tumor cells at 1:1 E:T ratio. Degranulation is relative to PMA positive control; representative normal donor is shown. Cytotoxic capacity of CAR-37 T cells was measured after overnight coculture with (E) Hut78 and (F) FEPD target cells at a different E:T ratio. The cytotoxicity assay is representative of 3 independent experiments conducted with different healthy donors. Mean ± SEM shown.
CAR-37 in vitro activity against T-cell lymphoma and leukemia. (A) CD37 expression on PTCL tumor cell lines. (B) Representative FACS plots from patient-derived samples. (C) CD69 expression and (D) CD107a degranulation of CAR T cells, as evaluated by flow cytometry after 6 hours of coculture with indicated tumor cells at 1:1 E:T ratio. Degranulation is relative to PMA positive control; representative normal donor is shown. Cytotoxic capacity of CAR-37 T cells was measured after overnight coculture with (E) Hut78 and (F) FEPD target cells at a different E:T ratio. The cytotoxicity assay is representative of 3 independent experiments conducted with different healthy donors. Mean ± SEM shown.
Bispecific CAR T cells against CD19 and CD37
A possible strategy to avoid antigen escape is to generate T cells capable of recognizing multiple antigens.25,26 A dual targeting of CD19 and CD37 has been previously investigated with a dual-ligand immunoliposomes approach, which has been shown to induce apoptosis of B-CLL cells.27 We designed a bispecific CAR that would efficiently trigger T-cell activation when CD19 or CD37 are present on the target cell alone or in combination. We generated and tested 2 constructs carrying anti-CD19 and anti-CD37 scFv’s connected in tandem in different orders in a second-generation 41BBz-CD3ζ vector (Figure 7A). We observed that the transduction efficiency was low for CAR-19-37 T cells compared with CAR37-19, even if the same MOI was used (supplemental Figure 5A-B).
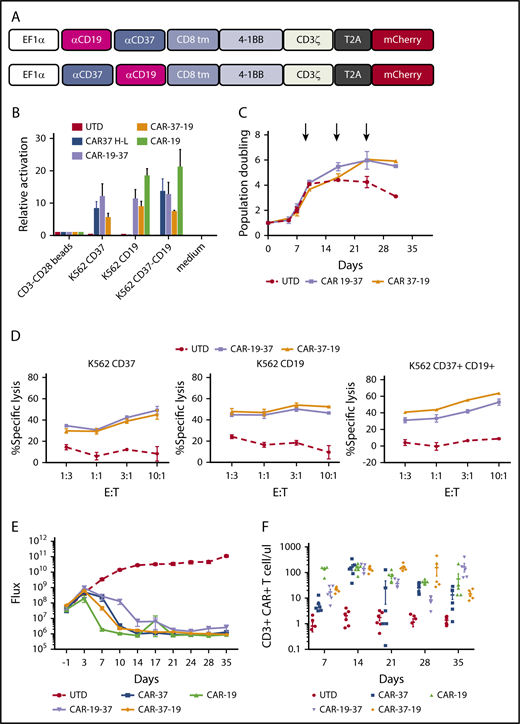
Bispecific CD19 CD37 CAR T cells. (A) Two bispecific second-generation CARs were constructed with a different order of the single-chain variable fragments: CAR-19-37 (top) and CAR-37-19 (bottom). (B) Activation of Jurkat reporter (NFAT-Luc) T cells transduced with different CAR constructs and cocultured with tumor cells. Luciferase activity was measured after 16 hours. CD3-CD28 beads: positive control. (C) Ex vivo expansion of CD3/CD28 bead-activated and target-stimulated T cells in 2 healthy donors for 30 days. (D) Cytotoxic capacity of bispecific CAR T cells was measured after overnight coculture with K562 targets transduced with CD37, CD19, or both at indicated E:T ratios. The cytotoxicity assay is representative of 2 independent experiments conducted with different healthy donors. (E) NSG mice were injected intravenously with 1 × 106 JEKO-1 (CBG−GFP) cells and monitored by BLI for tumor burden over time. At day 0, mice were randomly assigned on the basis of tumor burden (BLI) to receive 2 × 106 control T cells (UTD), CAR- 37, CAR-19, CAR-19-37, or CAR-37-19. All CAR T-cell groups were normalized to have the same % CAR+ cells and untransduced cells. Average flux (photons/s) of whole mice at different points is shown. Graph shows 1 experiment with 6 mice per group. (F) CAR T cells were enumerated in peripheral blood by flow cytometry at the indicated points. Absolute counts of CAR T cells are shown as mean ± SEM.
Bispecific CD19 CD37 CAR T cells. (A) Two bispecific second-generation CARs were constructed with a different order of the single-chain variable fragments: CAR-19-37 (top) and CAR-37-19 (bottom). (B) Activation of Jurkat reporter (NFAT-Luc) T cells transduced with different CAR constructs and cocultured with tumor cells. Luciferase activity was measured after 16 hours. CD3-CD28 beads: positive control. (C) Ex vivo expansion of CD3/CD28 bead-activated and target-stimulated T cells in 2 healthy donors for 30 days. (D) Cytotoxic capacity of bispecific CAR T cells was measured after overnight coculture with K562 targets transduced with CD37, CD19, or both at indicated E:T ratios. The cytotoxicity assay is representative of 2 independent experiments conducted with different healthy donors. (E) NSG mice were injected intravenously with 1 × 106 JEKO-1 (CBG−GFP) cells and monitored by BLI for tumor burden over time. At day 0, mice were randomly assigned on the basis of tumor burden (BLI) to receive 2 × 106 control T cells (UTD), CAR- 37, CAR-19, CAR-19-37, or CAR-37-19. All CAR T-cell groups were normalized to have the same % CAR+ cells and untransduced cells. Average flux (photons/s) of whole mice at different points is shown. Graph shows 1 experiment with 6 mice per group. (F) CAR T cells were enumerated in peripheral blood by flow cytometry at the indicated points. Absolute counts of CAR T cells are shown as mean ± SEM.
To determine whether bispecific CAR T cells could be activated in response to either antigen, we generated CAR-19, CAR-37, bispecific CAR19-37, and CAR37-19 Jurkat NFAT-Luc reporter cell lines and analyzed activation after overnight coculture with target cells. Bispecific CAR T cells showed high NFAT activation when either a single or both antigens were present on the surface of the target cells, and there was no discernible difference in activation signal in bispecific CAR T cells when they were activated by CD19 alone or CD37 alone (Figure 7B). Bispecific CAR T cells displayed comparable expansion after initial priming with anti-CD3/CD28 beads during the first 10 days (Figure 7C). Similarly, we found that bispecific CAR T cells could undergo long-term expansion through repetitive antigen stimulations, with K562 expressing CD19 and CD37 (Figure 7C). In a cytotoxicity assay with primary human CAR T cells, we found that bispecific CART cells responded to either a single target or both targets, and there was no discernible difference in cytotoxicity to CD19 or CD37 (Figure 7D). Last, we tested the bispecific constructs in vivo against Jeko-1 tumor cells, comparing them with conventional CAR-37 and CAR-19 T cells. By day 14 after treatment, there was complete disease eradication in all groups except CAR-19-37, which showed partial disease control (Figure 7E). This divergency may be related to structural differences between the 2 antigens and the order of the scFvs in the CAR constructs. We confirmed CAR T-cell persistence in the peripheral blood by flow cytometry through day 35 of treatment (Figure 7F). There was no discernible difference among CAR-37, CAR-19, and the optimal tumor-clearing bispecific CAR 37-19 at all of the points examined. Interestingly, the suboptimal CAR 19-37 had greater persistence at day 35, which may reflect persistent antigen stimulation by tumor. Taken together, these data indicate that bispecific CAR T cells caused specific target cytolysis of cells expressing 1 or both antigens on the surface, and that they are as effective as either monospecific CAR alone against MCL tumor in vivo.
Discussion
Adoptive cell therapy has progressed remarkably in the last decade, and impressive clinical responses have been obtained with anti-CD19 CAR T cells in patients with relapsed or refractory CD19+ leukemia and lymphomas.2-11,28 These data have yet to be recapitulated with other antigen targets for lymphoma, and some patients treated with CD19-directed therapies have relapsed with CD19-negative disease. In addition, patients with T-cell lymphoma are not candidates for CD19-directed CAR T cells. Thus, there is a need and desire to validate CAR T-cell therapies directed to alternative antigens.
CD37 is 1 of several developmentally regulated antigens expressed on mature human B cells and human B-cell malignancies. Expression data show that its presence in normal tissues is restricted to lymphoid organs including spleen, tonsil, lymph nodes, and bone marrow,18,29-31 and that it is expressed predominantly on mature B cells.17 CD37 is consistently expressed across most B-cell malignancies.17,23,32 In DLBCL, the most common type of NHL, CD37 has been reported to be expressed in nearly half of the cases,33,34 and most interestingly, CD37 is also expressed in some T-cell malignancies, but given the rarity of this disease subset, the frequency of expression has not been definitively determined. These reports suggest that optimal clinical trials targeting the CD37 antigen will include screening for CD37 as part of the patient selection, which is not current practice for CD19-targeted therapies. Nevertheless, screening is readily undertaken, and CD37 offers the possibility of treating both B- and T-cell malignancies. Encouragingly, recent clinical trials have shown the safety and efficacy of anti-CD37 immunotherapeutics. In particular, the monoclonal anti-CD37 antibody BI836826 and the humanized monospecific protein therapeutic otlertuzumab showed prolonged progression-free survival alone and in combination with other agents for the treatment of relapse/refractory CLL.24,35,36 Similarly, IMGN529, an immunoconjugate used for the treatment of non-Hodgkin lymphoma, showed safety and preliminary evidence of activity in patients with DLBCL.37,38 In a recent abstract presentation at the International Conference on Malignant Lymphoma in Lugano, Switzerland, the AGS67E antibody-drug conjugate directed to CD37 was found to be well tolerated in a phase 1 dose escalation trial, with neutropenia as the dose-limiting toxicity. The AGS67E drug showed single-agent activity in 16 (30%) of 53 patients with heavily pretreated non-Hodgkin lymphoma, including a partial response in 1 of 2 patients with cutaneous T-cell lymphoma, and partial responses in 2 of 4 patients with peripheral T-cell lymphoma. Some patients, including a patient with multiply relapsed mycosis fungoides, continued receiving the drug with a deep partial response for more than 2 years.39 Thus, given the expression profile in normal tissues and in B- and T-cell lymphomas, along with clinical safety data validating it as a target antigen, we chose to test and develop CAR T cells directed to CD37.
In this study, we demonstrated that T cells engineered with a second-generation CAR targeting CD37 antigen are potent therapeutically and can eradicate CD37-expressing tumors without causing significant T-cell fratricide or in vitro toxicity against other immune cells. This encouraging activity profile may be a result of the binding properties of the particular scFv we have integrated in our constructs, which may differ from other antibodies, many of which recognize glycosylated epitopes on CD37. We found that 1 configuration of the anti-CD37 constructs (CAR-37 H-L) was more effective than the other (CAR-37 L-H), according to cytolysis of multiple cell lines in vitro and tumor eradication of a MCL model in vivo. The 2 constructs differed by the orientation of the variable heavy and light chains of the scFv, but they bear the same intracellular domains of the CAR-19 construct we used as control reference (ie, 4-1BB and CD3 ζ). Notably, we observed disease control by CAR-37 H-L comparable to a CAR-19 T-cell treatment in a PDX model of MCL.
Relapse with CD19-negative disease after treatment with CD19-direted CAR T cells has been reported in different type of tumors, with an incidence exceeding 10% in patients with ALL.10,40,41 Therefore, we hypothesized that the long-term efficacy of immunotherapy could be improved if alternative and/or multiple antigens are targeted. In particular, targeting a novel antigen independent of CD19 may overcome potential antigen escape and offer a non-cross-resistant mechanism of antitumor activity. To this end, we also have explored the possibility of using anti-CD37 and anti-CD19 bispecific CAR T cells as a strategy to reduce antigen-loss tumor escape. Moreover, not all patients respond to CD19 CARs, despite continued expression of CD19 on their tumor. Although the mechanism of this resistance to CAR T-cell therapy is often thought to be related to inhibitory checkpoints, the role of the antigen and the antigen-specificity of the CAR have not yet been explored. Targeting an alternative antigen such as CD37 in this context would be 1 way of testing the hypothesis that the antigen specificity of CARs has a role in determining antitumor efficacy.
Finally, in addition to B-cell NHL, we and others19 have detected CD37 expression in primary patient PTCL samples. However, not all the PTCL lines or primary samples tested were CD37 positive, indicating that CART-37 is likely to be useful for only some PTCL cases, underlining the importance of screening for CD37 expression in tumors of patients considering CD37-targeted therapy. Importantly, targeting CD37+ PTCL is a strategy that does not require elimination of the healthy T-cell compartment, unlike many of the other strategies that have been proposed.22,42 In addition, some of the current strategies to target T-cell leukemias have focused on antigens that are only present in highly immature T cells, and are more relevant to T-cell acute lymphoblastic leukemia than for mature T-cell malignancies.
In summary, CD37 represents a promising target for the treatment of NHL and PTCL. A clinical trial of anti-CD37 CAR T cells is expected to open in 2019.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Katy Rezvani (University of Texas Anderson Cancer Center) and the Anderson CLL Moonshot Program for providing CLL samples. The authors also wish to thank Nora Horick for providing biostatistics advice, and Jennifer Guerriero for advice on macrophage differentiation.
I.S. receives support from American-Italian Cancer Foundation Post-Doctoral Research Fellowship. M.V.M. receives support from the Gabrielle’s Angel Foundation, the V Foundation, and the National Institutes of Health, National Cancer Institute (K08CA166039). D.M.W. and J.C.A. are supported by a Leukemia and Lymphoma Society Specialized Center of Research.
Authorship
Contribution: I.S., M.O., M.J.F., A.P.C., S.L., A.A.B., A.v.S., S.J.R., and A.J.S. performed the experiments; I.S. analyzed the results and created the figures; I.S. and M.V.M. designed the research; J.C.A. and F.I.P provided critical expertise in pathology and clinical flow cytometry, respectively; I.S. and M.V.M. wrote the manuscript; D.M.W. provided critical reagents and expertise and PDX models; and all authors read, edited, and approved the manuscript.
Conflict-of-interest disclosure: I.S. and M.V.M. are listed as inventors on patents related to this work and held by the Massachusetts General Hospital and Partners Health Care. The remaining authors declare no competing financial interests.
Correspondence: Marcela V. Maus, 149 13th St, Room 3.216, Charlestown, MA 02129; e-mail: mvmaus@mgh.harvard.edu.