

Key Points
Oral lenalidomide plus obinutuzumab is well tolerated and effective in patients with R/R FL.
The recommended dose of lenalidomide in combination with obinutuzumab 1000 mg was established as 20 mg.

Abstract
Obinutuzumab is a type II anti-CD20 monoclonal antibody that enhances antibody-dependent cellular cytotoxicity better than rituximab. Given promising results with lenalidomide and rituximab, this phase 1b study assessed the safety and efficacy of lenalidomide combined with obinutuzumab (GALEN). Patients age ≥18 years with relapsed or refractory (R/R) follicular lymphoma (FL) after rituximab-containing therapy received escalating doses (10 [n = 7], 15 [n = 3], 20 [n = 6], and 25 mg [n = 3]) of daily oral lenalidomide on days 1 to 21 of cycle 1 and on days 2 to 22 of cycles 2 to 6 (28-day cycles). Obinutuzumab 1000 mg IV was administered on days 8, 15, and 22 (cycle 1) and on day 1 (cycles 2-6). Dose was escalated in a 3 + 3 design based on dose-limiting toxicity (DLT) during cycle 1 to establish the maximum tolerated dose (MTD). We observed 164 adverse events (AEs), of which 139 were grade 1/2. The most common AEs were constipation (52.6%), neutropenia (47.4%), and asthenia (36.8%); 64.3% (9 of 14) of the grade 3/4 AEs were neutropenia/neutrophil decrease, but without any febrile neutropenia. Four DLTs occurred in 2 patients, all deemed unrelated to treatment. MTD was not reached. Twelve patients (63.2%) responded: 8 complete, 3 unconfirmed complete, and 1 partial response. Oral lenalidomide plus obinutuzumab is well tolerated and effective in R/R FL. The recommended dose of lenalidomide was established at 20 mg based on the risk of grade 3/4 neutropenia from cycle 2. This trial was registered at www.clinicaltrials.gov as #NCT01582776.
Introduction
Some of the most recent advances for the treatment of non-Hodgkin lymphoma (NHL) involve chemotherapy-free combinations as alternatives to immunochemotherapeutic regimens. Lenalidomide exerts direct immunomodulatory activity on lymphoma cells, enhances the function of T cells and natural killer (NK) cells, and improves antibody-dependent cellular cytotoxicity (ADCC) and antibody-dependent cellular phagocytosis.1 The actions of lenalidomide combined with the CD20 type I antibody rituximab have been shown to be synergistic in preclinical lymphoma models2-5 and effective in patients with various types of NHL6-11 in first-line6-8 and relapsed or refractory (R/R) settings.9-11
Obinutuzumab is a glycoengineered type II anti-CD20 monoclonal antibody (binding to a CD20 extracellular domain epitope overlapping with rituximab binding12 ) that enhances ADCC/antibody-dependent cellular phagocytosis and induces direct B-cell killing effects better than rituximab in preclinical models13,14 ; it has shown efficacy in NHL.15-18 We recently demonstrated that lenalidomide also triggered NK cell activation in vivo and that this effect was further improved upon subsequent obinutuzumab infusion, thereby enhancing directly and indirectly the efficacy of obinutuzumab.19 Thus, the combination of obinutuzumab and lenalidomide may be even more effective than rituximab plus lenalidomide. In 2012, a phase 1b/2 study was initiated to assess the safety and efficacy of obinutuzumab combined with lenalidomide (GALEN) for patients with R/R follicular lymphoma (FL).
Here, we report results of the phase 1b study, in which the primary objectives were to establish the recommended phase 2 dose (RP2D) of lenalidomide in combination with a fixed dose of obinutuzumab and to investigate the safety, tolerability, and preliminary antitumor activity of the combination in patients with R/R FL. Of note, the treatment schedule included 1 week of lenalidomide alone before the first obinutuzumab infusion, allowing separate evaluation of T-cell activation and CD20 modulation induced by lenalidomide from those related to the combination.
Patients and methods
Study design and patients
We performed a phase 1b, multicenter, open-label study sponsored by the Lymphoma Study Association using a 3 + 3 dose-escalation design to establish the maximum tolerated dose (MTD) of lenalidomide combined with obinutuzumab for patients with R/R FL. Patients were enrolled from 7 centers in France affiliated with the Lymphoma Study Association. The central independent ethics committee and the Agence Nationale de Sécurité du Médicament et des Produits de Santé approved the protocol, and the study was conducted in accordance with the ethical principles of the Declaration of Helsinki, Good Clinical Practices, and applicable regulatory requirements. All patients provided written informed consent.
Eligible patients were ≥18 years of age with a histopathologically confirmed diagnosis of CD20+ FL (World Health Organization grade 1, 2, or 3a) who had R/R disease after ≥1 systemic treatment containing rituximab and life expectancy ≥3 months. Additional inclusion criteria were an Eastern Cooperative Oncology Group performance status score of 0 to 2; adequate bone marrow, liver, and kidney function; and ≥1 bidimensionally measurable lesion on computed tomography (CT) scan (greatest transverse diameter >15 mm and short axis ≥10 mm). All patients were required to fulfill the lenalidomide requirements for pregnancy prevention.
The main exclusion criteria were central nervous system or leptomeningeal involvement by lymphoma, prior treatment with obinutuzumab or lenalidomide, and known CD20− status at relapse/progression. Patients were excluded if they had known infection with HIV, positive serology for hepatitis B or C, any serious active disease or comorbid medical condition (eg, severe cardiac disease), or any laboratory abnormalities not due to underlying lymphoma (eg, absolute neutrophil count <1.5 × 109/L, platelet count <100 × 109/L, aspartate aminotransferase or alanine aminotransferase ≥3.0× upper limit of normal, serum total bilirubin >34 µmol/L, or calculated creatinine clearance <50 mL/min). Patients were excluded if they had a history of other malignancies within 5 years (except for nonmelanoma skin tumors), any anticancer drug therapy within 28 days of initiation, or any corticosteroids within 4 weeks (except prednisone ≤10 mg/d).
Treatments
To identify the RP2D of lenalidomide, 4 patient cohorts received escalating doses (10, 15, 20, and 25 mg) of daily oral lenalidomide (on days 1-21 of the first 28-day cycle and on days 2-22 of 28-day cycles 2-6) in combination with IV infusions of obinutuzumab at a fixed dose of 1000 mg (on days 8, 15, and 22 of the first cycle and on day 1 of cycles 2-6; total of 8 infusions; Figure 1). Steroid premedication was mandatory before the first obinutuzumab infusion. All patients were required to take daily aspirin (100 mg) for prophylaxis against deep vein thrombosis (DVT) during the study period. Patients who could not tolerate aspirin, had a history of DVT, or had high risk of DVT received low–molecular weight heparin or warfarin. Growth factors were allowed in the study and recommended for up to 3 days if grade 4 neutropenia occurred, but they were not prophylactically administered.
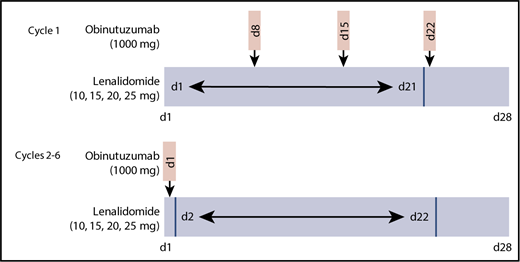
Treatment schedule. Escalating doses of oral lenalidomide (10, 15, 20, or 25 mg) were administered to 4 patient cohorts (n = 3-6) from days 1 to 21 in cycle 1 and day 2 to 22 in cycles 2 through 6. Obinutuzumab (1000 mg) IV was administered on days 8, 15, and 22 of cycle 1 and day 1 of cycles 2 through 6 (total of 8 infusions). Cycles are 28 days in length.
Treatment schedule. Escalating doses of oral lenalidomide (10, 15, 20, or 25 mg) were administered to 4 patient cohorts (n = 3-6) from days 1 to 21 in cycle 1 and day 2 to 22 in cycles 2 through 6. Obinutuzumab (1000 mg) IV was administered on days 8, 15, and 22 of cycle 1 and day 1 of cycles 2 through 6 (total of 8 infusions). Cycles are 28 days in length.
The lenalidomide dose was escalated in a 3 + 3 design based on the dose-limiting toxicity (DLT) assessment during cycle 1. The MTD was defined as the dose level before that which resulted in ≥2 of 6 patients experiencing DLTs. The RP2D was to be the MTD, but if the MTD was not reached, then the recommended dose was to be defined by all investigators according to the overall safety profile, approved by the data safety monitoring committee, and validated in 6 patients.
Objectives and outcomes
The primary objective was to establish the RP2D of lenalidomide in combination with a fixed dose of obinutuzumab. Secondary objectives included safety and efficacy of the therapeutic combination. Efficacy variables included complete response (CR), CR unconfirmed (CRu), partial response (PR), overall response rate (ORR; or CR/CRu/PR), CR rate (CRR; or CR/CRu), best ORR, progression-free survival (PFS), duration of response (DOR), and overall survival (OS) after 3 and 6 cycles according to the International Working Group (IWG) 1999 and IWG 2007).20,21 Patient response was assessed by the principal investigator.
Safety.
Safety evaluations included all adverse events (AEs) according to system organ class preferred term, clinical laboratory tests, vital sign measurements, physical examinations, electrocardiograms, and Eastern Cooperative Oncology Group performance status score. The National Cancer Institute Common Terminology Criteria for Adverse Events (version 4.03) were used to grade toxicity. Hematologic DLT was defined as grade ≥3 neutropenia or thrombocytopenia lasting ≥7 days, grade 4 neutropenia or thrombocytopenia lasting >3 days, or no recovery of absolute neutrophil count ≥1.5 × 109/L or platelet count ≥100 × 109/L by 8 weeks after the start of the previous cycle. Nonhematologic toxicity was defined as grade 3 blistering rash, grade 3 rash not resolving within 7 days after standard treatment, or grade 4 rash; grade 3 venous thrombosis/embolism despite adequate prevention; any grade 4 infusion-related reaction (IRR) during or within 24 hours after start of the first obinutuzumab infusion or any grade 3 IRR not resolving to grade 2 despite intervention (reduced infusion rate, temporary stop, supportive care, and/or corticosteroids); any grade 3 tumor flare reaction (TFR) or grade 3 TFR not resolving within 5 days after standard medical treatment; or any other grade ≥3 nonhematologic toxicity. Patients who experienced a DLT during the first cycle were not replaced. Patients who discontinued the first cycle without experiencing safety problems were replaced to ensure 3 safety-evaluable patients per dose cohort.
Efficacy.
Patients were evaluated using CT scans with IV contrast of the neck, chest, abdomen, and pelvis and with whole-body positron emission tomography (PET) scans. Disease response was assessed by the Revised Response Criteria for Malignant Lymphoma (IWG 1999 and IWG 2007).20,21 Tumor assessments (clinical examination, laboratory tests, abdominal and chest CT scans, PET scan, and bone marrow examination) were performed at baseline, after cycle 3, and 4 weeks after cycle 6 (except bone marrow examination if negative at baseline). During follow-up, clinical and neurologic examinations and laboratory tests were repeated every 3 months, and imaging studies (CT; optional PET) were repeated every 6 months.
Exploratory end points.
The study included a pilot exploration of some specific aspects of the biologic impact of the treatment strategy on malignant cells and immune response. Heparinized blood was drawn at various time points to perform a thorough analysis of peripheral blood immune-cell subsets (phenotyping of T cells, malignant and normal B cells, NK cells, and myeloid cells) by flow cytometry before and 1 week after lenalidomide treatment, shortly after the first obinutuzumab infusion, and at the end of 1 and 6 cycles of treatment.
Statistical analysis
The sample size estimate for this phase 1b study was based on a 3 + 3 escalation rule with 4 dose levels and therefore needed to include between 3 and 24 patients. Safety evaluations were summarized descriptively. The treated population consisted of all patients who received ≥1 dose of the study drug. This population was used for all safety and efficacy analyses. Response rate 95% confidence intervals (CIs) were calculated according to the exact Pearson-Clopper method. Survival functions were estimated using Kaplan-Meier methodology with appropriate 95% CIs. Immune parameters measured as continuous values were compared by Wilcoxon matched-pairs signed-rank test using GraphPad Prism 5.0 software.
Results
Patient disposition and demographics
Twenty patients with FL were enrolled between October 2012 and January 2014. Patients were to receive daily lenalidomide 10 (n = 7), 15 (n = 3), 20 (n = 6), or 25 mg (n = 4). One patient enrolled at 25 mg was withdrawn before receiving any treatment because of neutropenia occurring at baseline screening, leaving 19 patients evaluable for safety and efficacy. A patient receiving 10 mg was not evaluable for DLT and was replaced because she missed 1 infusion of obinutuzumab during the first cycle as a result of fortuitous concomitant diagnosis of breast cancer at day 4 on microbiopsies of breast lesions that were presumed to be lymphomatous (n = 18 for DLT assessment). Two patients permanently discontinued the study because of AEs, including cardiac arrest at cycle 1 (n = 1), ischemic stroke at cycle 3 (n = 1); disease progression (n = 2); and study treatment toxicity (n = 1). Median number of treatment cycles was 6 (range, 1-6). Median percentage of planned doses (total dose taken × 100/total dose expected in milligrams) taken for both lenalidomide and obinutuzumab was ≥90%.
Half of the enrolled patients were men, and the mean age of all patients was 61.5 years (Table 1). Patients mostly had Ann Arbor stage IV disease (70%) and had received 1 to 5 prior systemic therapies (median, 2). Four patients were refractory to their last lymphoma therapy, and 8 were refractory to rituximab (Table 1).
Demographic and disease characteristics in the enrolled set (N = 20)
| Characteristic | N (%) |
|---|---|
| Age, y | |
| Mean | 61.5 |
| Standard deviation | 12.5 |
| Range | 39-80 |
| Sex | |
| Male | 10 (50) |
| Female | 10 (50) |
| Initial histology (local) | |
| CD20+ FL grade 1 | 9 (45) |
| CD20+ FL grade2 | 7 (35) |
| CD20+ FL grade 3a | 1 (5) |
| CD20+ FL (grade unknown) | 3 (15) |
| No. of prior treatments | |
| Median | 2.0 |
| Range | 1-5 |
| Ann Arbor stage | |
| 1-2 | 3 (15) |
| 3-4 | 17 (85) |
| Performance status (ECOG) | |
| 0 | 16 (80) |
| 1 | 4 (20) |
| Bone marrow involvement | 6 (40)* |
| LDH, IU/L | |
| Normal | 13 (65) |
| >Upper limit of normal | 7 (35) |
| Bulk >5 cm | |
| No | 15 (75) |
| Yes | 5 (25) |
| Calculated FLIPI score | |
| 0 | 2 (10) |
| 1 | 3 (15) |
| 2 | 6 (30) |
| 3 | 7 (35) |
| 4 | 1 (5) |
| 5 | 1 (5) |
| Refractory to rituximab | |
| No | 12 (60) |
| Yes | 8 (40) |
| Refractory to prior lymphoma therapy | |
| No | 16 (80) |
| Yes | 4 (20) |
| Characteristic | N (%) |
|---|---|
| Age, y | |
| Mean | 61.5 |
| Standard deviation | 12.5 |
| Range | 39-80 |
| Sex | |
| Male | 10 (50) |
| Female | 10 (50) |
| Initial histology (local) | |
| CD20+ FL grade 1 | 9 (45) |
| CD20+ FL grade2 | 7 (35) |
| CD20+ FL grade 3a | 1 (5) |
| CD20+ FL (grade unknown) | 3 (15) |
| No. of prior treatments | |
| Median | 2.0 |
| Range | 1-5 |
| Ann Arbor stage | |
| 1-2 | 3 (15) |
| 3-4 | 17 (85) |
| Performance status (ECOG) | |
| 0 | 16 (80) |
| 1 | 4 (20) |
| Bone marrow involvement | 6 (40)* |
| LDH, IU/L | |
| Normal | 13 (65) |
| >Upper limit of normal | 7 (35) |
| Bulk >5 cm | |
| No | 15 (75) |
| Yes | 5 (25) |
| Calculated FLIPI score | |
| 0 | 2 (10) |
| 1 | 3 (15) |
| 2 | 6 (30) |
| 3 | 7 (35) |
| 4 | 1 (5) |
| 5 | 1 (5) |
| Refractory to rituximab | |
| No | 12 (60) |
| Yes | 8 (40) |
| Refractory to prior lymphoma therapy | |
| No | 16 (80) |
| Yes | 4 (20) |
ECOG, Eastern Cooperative Oncology Group; FLIPI, Follicular Lymphoma International Prognostic Index; LDH, lactic dehydrogenase.
n = 15.
DLT
Four DLTs occurred in 2 of the 18 evaluable patients during cycle 1: 1 death resulting from cardiac arrest at 10 mg in the presence of grade 3 worsening pleural effusion, and another patient treated at 20 mg who had grade 3 pulmonary infection with grade 3 hypokalemia; both cases were deemed unrelated to study treatment. Although the MTD was not reached, the toxicity review committee suggested an RP2D of 20 mg of lenalidomide instead of 25 mg in view of the incidence of grade 3 and 4 neutropenias during cycle 2 at 25 mg (Table 2), which may have clinical relevance or an impact on treatment exposure during phase 2. An additional 3 patients were enrolled in the 20-mg cohort, confirming the tolerability of this dosage. The suggested RP2D was validated by the data safety monitoring committee.
AEs occurring in ≥10% of patients and all AEs of grade 3 or 4 in treated set by dose group
| AE | N (%) | ||||
|---|---|---|---|---|---|
| Lenalidomide dose, mg | Treated set (N = 19) | ||||
| 10 (n = 7) | 15 (n = 3) | 20 (n = 6) | 25 (n = 3) | ||
| Total with ≥1 AE (all grades) | 7 (100) | 3 (100) | 6 (100) | 3 (100) | 19 (100) |
| Constipation | 3 (42.9) | 1 (33.3) | 5 (83.3) | 1 (33.3) | 10 (52.6) |
| Neutropenia/decreased neutrophil count | 1 (14.3) | 1 (33.3) | 4 (66.7) | 3 (100) | 9 (47.4) |
| Asthenia | 1 (14.3) | 2 (66.7) | 2 (33.3) | 2 (66.7) | 7 (36.8) |
| Cough | 1 (14.3) | 2 (66.7) | 1 (16.7) | 1 (33.3) | 5 (26.3) |
| Muscle spasms | 1 (14.3) | 0 (0.0) | 3 (50.0) | 1 (33.3) | 5 (26.3) |
| Diarrhea | 1 (14.3) | 1 (33.3) | 1 (16.7) | 1 (33.3) | 4 (21.1) |
| Pyrexia | 2 (28.6) | 1 (33.3) | 1(16.7) | 0 (0.0) | 4 (21.1) |
| Infusion-related reaction | 0 (0.0) | 1 (33.3) | 1 (16.7) | 1 (33.3) | 3 (15.8) |
| Nausea | 1 (14.3) | 1 (33.3) | 1 (16.7) | 0 (0.0) | 3 (15.8) |
| Rash | 1 (14.3) | 1 (33.3) | 1 (16.7) | 0 (0.0) | 3 (15.8) |
| Rhinitis, allergic | 0 (0.0) | 1 (33.3) | 1 (16.7) | 1 (33.3) | 3 (15.8) |
| Weight increase | 1 (14.3) | 1 (33.3) | 0 (0.0) | 1 (33.3) | 3 (15.8) |
| Weight loss | 2 (28.6) | 0 (0.0) | 1 (16.7) | 0 (0.0) | 3 (15.8) |
| Bronchitis | 1 (14.3) | 1 (33.3) | 0 (0.0) | 0 (0.0) | 2 (10.5) |
| Dry skin | 0 (0.0) | 1 (33.3) | 1 (16.7) | 0 (0.0) | 2 (10.5) |
| Fatigue | 1 (14.3) | 0 (0.0) | 0 (0.0) | 1 (33.3) | 2 (10.5) |
| Headache | 0 (0.0) | 1 (33.3) | 0 (0.0) | 1 (33.3) | 2 (10.5) |
| Rhinitis | 0 (0.0) | 0 (0.0) | 1 (16.7) | 1 (33.3) | 2 (10.5) |
| Sinusitis | 0 (0.0) | 0 (0.0) | 2 (33.3) | 0 (0.0) | 2 (10.5) |
| Upper abdominal pain | 2 (28.6) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 2 (10.5) |
| Urinary tract infection | 0 (0.0) | 1 (33.3) | 0 (0.0) | 1 (33.3) | 2 (10.5) |
| Total with ≥1 grade 3/4 AE | 4 (57.1) | 2 (66.7) | 5 (83.3) | 3 (100) | 14 (73.7) |
| Neutropenia/decreased neutrophil count | 1 (14.3) | 1 (33.3) | 4 (66.7) | 3 (100) | 9 (47.4) |
| Anemia | 0 (0.0) | 0 (0.0) | 1 (16.7) | 0 (0.0) | 1 (5.3) |
| Acute myeloid leukemia | 0 (0.0) | 1 (33.3) | 0 (0.0) | 0 (0.0) | 1 (5.3) |
| Breast cancer | 1 (14.3) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (5.3) |
| Hypertension | 1 (14.3) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (5.3) |
| Hypokalemia | 0 (0.0) | 0 (0.0) | 1 (16.7) | 0 (0.0) | 1 (5.3) |
| Ischemic stroke | 0 (0.0) | 0 (0.0) | 1 (16.7) | 0 (0.0) | 1 (5.3) |
| Lung infection | 0 (0.0) | 0 (0.0) | 1 (16.7) | 0 (0.0) | 1 (5.3) |
| Pleural effusion | 1 (14.3) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (5.3) |
| Pulmonary embolism | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (33.3) | 1 (5.3) |
| AE | N (%) | ||||
|---|---|---|---|---|---|
| Lenalidomide dose, mg | Treated set (N = 19) | ||||
| 10 (n = 7) | 15 (n = 3) | 20 (n = 6) | 25 (n = 3) | ||
| Total with ≥1 AE (all grades) | 7 (100) | 3 (100) | 6 (100) | 3 (100) | 19 (100) |
| Constipation | 3 (42.9) | 1 (33.3) | 5 (83.3) | 1 (33.3) | 10 (52.6) |
| Neutropenia/decreased neutrophil count | 1 (14.3) | 1 (33.3) | 4 (66.7) | 3 (100) | 9 (47.4) |
| Asthenia | 1 (14.3) | 2 (66.7) | 2 (33.3) | 2 (66.7) | 7 (36.8) |
| Cough | 1 (14.3) | 2 (66.7) | 1 (16.7) | 1 (33.3) | 5 (26.3) |
| Muscle spasms | 1 (14.3) | 0 (0.0) | 3 (50.0) | 1 (33.3) | 5 (26.3) |
| Diarrhea | 1 (14.3) | 1 (33.3) | 1 (16.7) | 1 (33.3) | 4 (21.1) |
| Pyrexia | 2 (28.6) | 1 (33.3) | 1(16.7) | 0 (0.0) | 4 (21.1) |
| Infusion-related reaction | 0 (0.0) | 1 (33.3) | 1 (16.7) | 1 (33.3) | 3 (15.8) |
| Nausea | 1 (14.3) | 1 (33.3) | 1 (16.7) | 0 (0.0) | 3 (15.8) |
| Rash | 1 (14.3) | 1 (33.3) | 1 (16.7) | 0 (0.0) | 3 (15.8) |
| Rhinitis, allergic | 0 (0.0) | 1 (33.3) | 1 (16.7) | 1 (33.3) | 3 (15.8) |
| Weight increase | 1 (14.3) | 1 (33.3) | 0 (0.0) | 1 (33.3) | 3 (15.8) |
| Weight loss | 2 (28.6) | 0 (0.0) | 1 (16.7) | 0 (0.0) | 3 (15.8) |
| Bronchitis | 1 (14.3) | 1 (33.3) | 0 (0.0) | 0 (0.0) | 2 (10.5) |
| Dry skin | 0 (0.0) | 1 (33.3) | 1 (16.7) | 0 (0.0) | 2 (10.5) |
| Fatigue | 1 (14.3) | 0 (0.0) | 0 (0.0) | 1 (33.3) | 2 (10.5) |
| Headache | 0 (0.0) | 1 (33.3) | 0 (0.0) | 1 (33.3) | 2 (10.5) |
| Rhinitis | 0 (0.0) | 0 (0.0) | 1 (16.7) | 1 (33.3) | 2 (10.5) |
| Sinusitis | 0 (0.0) | 0 (0.0) | 2 (33.3) | 0 (0.0) | 2 (10.5) |
| Upper abdominal pain | 2 (28.6) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 2 (10.5) |
| Urinary tract infection | 0 (0.0) | 1 (33.3) | 0 (0.0) | 1 (33.3) | 2 (10.5) |
| Total with ≥1 grade 3/4 AE | 4 (57.1) | 2 (66.7) | 5 (83.3) | 3 (100) | 14 (73.7) |
| Neutropenia/decreased neutrophil count | 1 (14.3) | 1 (33.3) | 4 (66.7) | 3 (100) | 9 (47.4) |
| Anemia | 0 (0.0) | 0 (0.0) | 1 (16.7) | 0 (0.0) | 1 (5.3) |
| Acute myeloid leukemia | 0 (0.0) | 1 (33.3) | 0 (0.0) | 0 (0.0) | 1 (5.3) |
| Breast cancer | 1 (14.3) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (5.3) |
| Hypertension | 1 (14.3) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (5.3) |
| Hypokalemia | 0 (0.0) | 0 (0.0) | 1 (16.7) | 0 (0.0) | 1 (5.3) |
| Ischemic stroke | 0 (0.0) | 0 (0.0) | 1 (16.7) | 0 (0.0) | 1 (5.3) |
| Lung infection | 0 (0.0) | 0 (0.0) | 1 (16.7) | 0 (0.0) | 1 (5.3) |
| Pleural effusion | 1 (14.3) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (5.3) |
| Pulmonary embolism | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (33.3) | 1 (5.3) |
Safety
All 19 patients (100%) experienced at least 1 AE; the total number of AEs was 164 (Table 2). The most common AEs (all grades, ≥20% of patients) were constipation (52.6%), neutropenia/ decreased neutrophil count (47.4%), asthenia (36.8%), cough (26.3%), muscle spasms (26.3%), diarrhea (21.1%), and pyrexia (21.1%). Grade 1 (50.6%) or 2 (34.1%) was the most common grade of AEs regardless of causality; AEs of worst grade 3, 4, and 5 represented 11.0%, 3.0%, and 1.2% of all AEs, respectively (Table 2).
Thirteen AEs (7.9%) were deemed related only to obinutuzumab, 54 (32.9%) were related only to lenalidomide, and 32 (19.5%) were related to the combination of both. Neutropenia was the only drug-related grade 3 or 4 AE occurring in >2 patients (Table 2), and none of these were febrile neutropenia. Other drug-related AEs of special interest, including IRRs (n = 3), rash (n = 3), and TFR (n = 1), were rare and mild (worst grade was 2).
A total of 9 serious AEs (SAEs) were recorded in 7 patients (36.8%), including 3 (33.3%) second primary malignancies and 4 (44.4%) occurring during cycle 1. Four SAEs were deemed related only to lenalidomide, including cerebral ischemic stroke at cycle 2, acute monocytic leukemia (World Health Organization), pulmonary embolism, and pancreatic carcinoma (n = 1 each). The other 5 SAEs were deemed unrelated to either study treatment and included pulmonary infection, cardiac arrest, costal fracture, worsening pleural effusion, and breast cancer (n = 1 each).
Six (31.6%) of 19 patients died during the study. Three patients died as a result of lymphoma, including a 70-year-old man at cycle 4 and 2 women during the follow-up period (age 39 and 43 years). A fourth patient had an unexplained death at day 14 of cycle 1; this 73-year-old man had worsening pleural effusion because of FL; he died as a result of cardiac arrest, presumably because of pulmonary embolism or cardiac dysrhythmia, which was considered unrelated to either treatment. The fifth patient was a 60-year-old man who died as a result of metastatic pancreatic carcinoma diagnosed 20 months after initiation of study treatment while still in CR to treatment for FL; he had received 4 prior regimens, including first-line consolidation with BEAM (carmustine, etoposide, cytarabine, and melphalan) followed by autologous stem-cell transplantation in 1992 and second-line therapy with fludarabine/mitoxantrone in 1998. The sixth patient was a 64-year-old man who died as a result of bilateral pneumopathy in the context of acute monocytic leukemia diagnosed 3 years after initiation of study treatment; he had received 4 prior therapies, including R-CHOP (rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone) and involved-field radiotherapy twice.
Efficacy
The 19 patients included in the safety set were evaluated for efficacy (patients with missing assessments were considered nonresponders). ORR and CRR after 3 cycles (intermediate assessment) and 6 cycles (end of treatment) according to IWG 1999 and 2007 are summarized in Table 3. Overall, after 6 cycles in the total safety set, OR (CR/CRu/PR) was achieved by 12 patients (63.2%; 95% CI, 38.4-83.7) according to both IWG 1999 and IWG 2007. CRR (CR/CRu) at the end of treatment was achieved by 11 patients (57.9%; 95% CI, 33.5-79.7). Seven patients experienced progression (n = 4) or relapse (n = 3) during the study.
RRs by IWG 1999 and 2007 criteria at intermediate and final assessments (response-evaluable patient population)
| Assessment time | N (%) | ||||
|---|---|---|---|---|---|
| Lenalidomide dose, mg | Total (N = 19) | ||||
| 10 (n = 7) | 15 (n = 3) | 20 (n = 6) | 25 (n = 3) | ||
| IWG 199920 | |||||
| Intermediate (3 mo) | |||||
| CR | 0 | 1 (33.3) | 2 (33.3) | 1 (33.3) | 4 (21.1) |
| CRu | 1 (14.3) | 0 | 0 | 0 | 1 (5.3) |
| PR | 2 (28.6) | 2 (66.7) | 2 (33.3) | 2 (66.7) | 8 (42.1) |
| SD | 3 (42.9) | 0 | 0 | 0 | 3 (15.8) |
| PD | 0 | 0 | 1 (16.7) | 0 | 1 (5.3) |
| Not evaluated | 1 (14.3) | 0 | 1 (16.7) | 0 | 2 (10.5) |
| CRR (CR/CRu) | 1 (14.3) | 1 (33.3) | 2 (33.3) | 1 (33.3) | 5 (26.3) |
| 95% CI | 9.1-51.2 | ||||
| ORR (CR/CRu/PR) | 3 (42.9) | 3 (100.0) | 4 (66.7) | 3 (100.0) | 13 (68.4) |
| 95% CI | 43.4-87.4 | ||||
| End of treatment | |||||
| CR | 1 (14.3) | 2 (66.7) | 3 (50.0) | 2 (66.7) | 8 (42.1) |
| CRu | 2 (28.6) | 1 (33.3) | 0 | 0 | 3 (15.8) |
| PR | 0 | 0 | 0 | 1 (33.3) | 1 (5.3) |
| SD | 0 | 0 | 0 | 0 | 0 |
| PD | 3 (42.9) | 0 | 2 (33.3) | 0 | 5 (26.3) |
| Not evaluated | 1 (14.3) | 0 | 1 (16.7) | 0 | 2 (10.5) |
| CRR (CR/CRu) | 3 (42.9) | 3 (100.0) | 3 (50.0) | 2 (66.7) | 11 (57.9) |
| 95% CI | 33.5-79.7 | ||||
| ORR (CR/CRu/PR) | 3 (42.9) | 3 (100.0) | 3 (50.0) | 3 (100.0) | 12 (63.2) |
| 95% CI | 38.4-83.7 | ||||
| IWG 200721 | |||||
| Intermediate (3 mo) | |||||
| CR | 1 (14.3) | 1 (33.3) | 2 (33.3) | 2 (66.7) | 6 (31.6) |
| PR | 2 (28.6) | 2 (66.7) | 3 (50.0) | 1 (33.3) | 8 (42.1) |
| SD | 2 (28.6) | 0 | 0 | 0 | 2 (10.5) |
| PD | 1 (14.3) | 0 | 1 (16.7) | 0 | 2 (10.5) |
| Not evaluated | 1 (14.3) | 0 | 0 | 0 | 1 (5.3) |
| CRR (CR/CRu) | 1 (14.3) | 1 (33.3) | 2 (33.3) | 2 (66.7) | 6 (31.6) |
| 95% CI | 12.6-56.6 | ||||
| ORR (CR/CRu/PR) | 3 (42.9) | 3 (100.0) | 5 (83.3) | 3 (100.0) | 14 (73.7) |
| 95% CI | 48.8-90.9 | ||||
| End of treatment | |||||
| CR | 3 (42.9) | 3 (100.0) | 2 (33.3) | 3 (100.0) | 11 (57.9) |
| PR | 0 | 0 | 1 (16.7) | 0 | 1 (5.3) |
| SD | 0 | 0 | 0 | 0 | 0 |
| PD | 2 (28.6) | 0 | 2 (33.3) | 0 | 4 (21.1) |
| Not evaluated | 2 (28.6) | 0 | 1 (16.7) | 0 | 3 (15.8) |
| CRR (CR/CRu) | 3 (42.9) | 3 (100.0) | 2 (33.3) | 3 (100.0) | 11 (57.9) |
| 95% CI | 33.5-79.7 | ||||
| ORR (CR/CRu/PR) | 3 (42.9) | 3 (100.0) | 3 (50.0) | 3 (100.0) | 12 (63.2) |
| 95% CI | 38.4-83.7 | ||||
| Assessment time | N (%) | ||||
|---|---|---|---|---|---|
| Lenalidomide dose, mg | Total (N = 19) | ||||
| 10 (n = 7) | 15 (n = 3) | 20 (n = 6) | 25 (n = 3) | ||
| IWG 199920 | |||||
| Intermediate (3 mo) | |||||
| CR | 0 | 1 (33.3) | 2 (33.3) | 1 (33.3) | 4 (21.1) |
| CRu | 1 (14.3) | 0 | 0 | 0 | 1 (5.3) |
| PR | 2 (28.6) | 2 (66.7) | 2 (33.3) | 2 (66.7) | 8 (42.1) |
| SD | 3 (42.9) | 0 | 0 | 0 | 3 (15.8) |
| PD | 0 | 0 | 1 (16.7) | 0 | 1 (5.3) |
| Not evaluated | 1 (14.3) | 0 | 1 (16.7) | 0 | 2 (10.5) |
| CRR (CR/CRu) | 1 (14.3) | 1 (33.3) | 2 (33.3) | 1 (33.3) | 5 (26.3) |
| 95% CI | 9.1-51.2 | ||||
| ORR (CR/CRu/PR) | 3 (42.9) | 3 (100.0) | 4 (66.7) | 3 (100.0) | 13 (68.4) |
| 95% CI | 43.4-87.4 | ||||
| End of treatment | |||||
| CR | 1 (14.3) | 2 (66.7) | 3 (50.0) | 2 (66.7) | 8 (42.1) |
| CRu | 2 (28.6) | 1 (33.3) | 0 | 0 | 3 (15.8) |
| PR | 0 | 0 | 0 | 1 (33.3) | 1 (5.3) |
| SD | 0 | 0 | 0 | 0 | 0 |
| PD | 3 (42.9) | 0 | 2 (33.3) | 0 | 5 (26.3) |
| Not evaluated | 1 (14.3) | 0 | 1 (16.7) | 0 | 2 (10.5) |
| CRR (CR/CRu) | 3 (42.9) | 3 (100.0) | 3 (50.0) | 2 (66.7) | 11 (57.9) |
| 95% CI | 33.5-79.7 | ||||
| ORR (CR/CRu/PR) | 3 (42.9) | 3 (100.0) | 3 (50.0) | 3 (100.0) | 12 (63.2) |
| 95% CI | 38.4-83.7 | ||||
| IWG 200721 | |||||
| Intermediate (3 mo) | |||||
| CR | 1 (14.3) | 1 (33.3) | 2 (33.3) | 2 (66.7) | 6 (31.6) |
| PR | 2 (28.6) | 2 (66.7) | 3 (50.0) | 1 (33.3) | 8 (42.1) |
| SD | 2 (28.6) | 0 | 0 | 0 | 2 (10.5) |
| PD | 1 (14.3) | 0 | 1 (16.7) | 0 | 2 (10.5) |
| Not evaluated | 1 (14.3) | 0 | 0 | 0 | 1 (5.3) |
| CRR (CR/CRu) | 1 (14.3) | 1 (33.3) | 2 (33.3) | 2 (66.7) | 6 (31.6) |
| 95% CI | 12.6-56.6 | ||||
| ORR (CR/CRu/PR) | 3 (42.9) | 3 (100.0) | 5 (83.3) | 3 (100.0) | 14 (73.7) |
| 95% CI | 48.8-90.9 | ||||
| End of treatment | |||||
| CR | 3 (42.9) | 3 (100.0) | 2 (33.3) | 3 (100.0) | 11 (57.9) |
| PR | 0 | 0 | 1 (16.7) | 0 | 1 (5.3) |
| SD | 0 | 0 | 0 | 0 | 0 |
| PD | 2 (28.6) | 0 | 2 (33.3) | 0 | 4 (21.1) |
| Not evaluated | 2 (28.6) | 0 | 1 (16.7) | 0 | 3 (15.8) |
| CRR (CR/CRu) | 3 (42.9) | 3 (100.0) | 2 (33.3) | 3 (100.0) | 11 (57.9) |
| 95% CI | 33.5-79.7 | ||||
| ORR (CR/CRu/PR) | 3 (42.9) | 3 (100.0) | 3 (50.0) | 3 (100.0) | 12 (63.2) |
| 95% CI | 38.4-83.7 | ||||
Response-evaluable patients were those who received ≥1 dose of study treatment and had baseline and ≥1 posttreatment tumor assessment. Patients with missing assessments were considered nonresponders.
PD, progressive disease; SD, stable disease.
Median follow-up for the final analysis was 38.1 months (95% CI, 35.0-41.6). At 3 years, PFS was 52.1% (95% CI, 28.0-71.6), DOR was 68.4% (95% CI, 35.9-86.8), and OS was 73.3% (95% CI, 47.2-87.9), illustrated in Figure 2A-C.
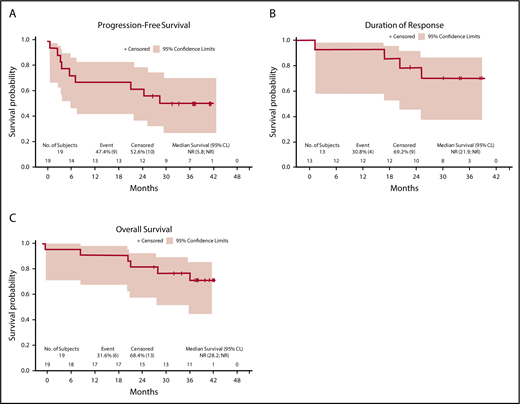
PFS, DOR, and OS in the treated set. (A) PFS; (B) DOR; and (C) OS. CL, confidence limit; NR, not reached.
PFS, DOR, and OS in the treated set. (A) PFS; (B) DOR; and (C) OS. CL, confidence limit; NR, not reached.
Biologic evaluation
As soon as 1 week after lenalidomide dosing, T lymphocytes were strongly activated in peripheral blood, as exemplified by the increase in the percentage of these cells expressing high levels of HLA-DR (HLA-DRbright; Figure 3A). Of note, HLA-DRbright T cells further increased 2 hours after obinutuzumab infusion. This activation was transient; HLA-DR expression returned to the basal level at the end of cycle 1 of treatment (ie, after a 1-week washout of lenalidomide). Nevertheless, some of the activated T cells persisted over time during treatment courses; HLA-DR expression remained significantly higher after 6 cycles (end of induction; Figure 3A). In parallel, we analyzed whether the obinutuzumab target was altered by concomitant lenalidomide. CD20 surface expression was not modulated in normal B cells. Moreover, 9 patients displaying detectable circulating malignant B cells (99%; of blood B cells; range, 14%-100%) also showed a lack of downregulation of CD20 on tumor cells (Figure 3B).
![Figure 3. Lenalidomide activates T cells in vivo and does not alter CD20 expression. (A) HLA-DR expression was measured by flow cytometry in peripheral blood on CD4 and CD8 T cells at various time points. (B) CD20 expression was measured by flow cytometry on circulating normal and/or malignant B cells. End of induction was 28th day of sixth cycle of treatment. Median value is depicted as a black bar. *P ≤ .05, ***P ≤ .001. C1D1, first day of first treatment cycle (before lenalidomide intake); C1D8, eighth day of first cycle (before or after obinutuzumab [GA101] infusion); C2D1, first day of second cycle (before lenalidomide intake); MFI, mean fluorescence intensity; ns, not significant.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/132/14/10.1182_blood-2018-05-853499/4/m_blood853499f3.png?Expires=1767702440&Signature=OTDRqP9GCmwMyL4eN1Md6r3UJM0MR8sYnJZqABrSyTpB20E0zZnyo8wDJIuZCgoMwrR9Vzw4nAoI5bdO3~G7jKVyiafOIzlnxEE1JpiqFLyJdoLnDOZwqQKGosv8KIkvzHJF7b5aSmf3e7HOHDF-rTgqdaZxmLOQVHv974lsBhSUNw-m5HXwWSVP0yrjvC-HaOREFbrxSX-vUu10V60N1aq7zfpHlsxezzkgi7qZx6P8A-TcyLvmC1BpxpW44J3woMQlFXG0i0kIPgZvnpS9cM~ybFlf27cgA6a2gmHjkmhN1LA5e9mzjorX-nz6y2~wZYYmL4AX~YsrNbmYqsVPtw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Lenalidomide activates T cells in vivo and does not alter CD20 expression. (A) HLA-DR expression was measured by flow cytometry in peripheral blood on CD4 and CD8 T cells at various time points. (B) CD20 expression was measured by flow cytometry on circulating normal and/or malignant B cells. End of induction was 28th day of sixth cycle of treatment. Median value is depicted as a black bar. *P ≤ .05, ***P ≤ .001. C1D1, first day of first treatment cycle (before lenalidomide intake); C1D8, eighth day of first cycle (before or after obinutuzumab [GA101] infusion); C2D1, first day of second cycle (before lenalidomide intake); MFI, mean fluorescence intensity; ns, not significant.
Lenalidomide activates T cells in vivo and does not alter CD20 expression. (A) HLA-DR expression was measured by flow cytometry in peripheral blood on CD4 and CD8 T cells at various time points. (B) CD20 expression was measured by flow cytometry on circulating normal and/or malignant B cells. End of induction was 28th day of sixth cycle of treatment. Median value is depicted as a black bar. *P ≤ .05, ***P ≤ .001. C1D1, first day of first treatment cycle (before lenalidomide intake); C1D8, eighth day of first cycle (before or after obinutuzumab [GA101] infusion); C2D1, first day of second cycle (before lenalidomide intake); MFI, mean fluorescence intensity; ns, not significant.
Discussion
In this first clinical study of combined obinutuzumab and lenalidomide (GALEN) administered for 6 months, an acceptable safety and tolerability profile was found in patients with R/R FL. Four DLTs were reported, the MTD was not reached, and most of the drug-related AEs were grade 1 or 2. Grade 3 or 4 hematopoietic AEs (primarily neutropenia/decreased neutrophil count) were observed as laboratory findings (without any cases of grade 3 or 4 febrile neutropenia) in 11 patients. There was no signal of increased incidence or more severe infusion-related reactions, thromboembolic events, or thrombocytopenia. Although 2 secondary primary malignancies were deemed possibly related, both patients had received heavy prior therapy that could have contributed to their emergence. The 20-mg dose was selected as the RP2D based on the safety profile, particularly the increasing incidence of grade 3 and 4 neutropenia at 25 mg after cycle 1.
In the small group of patients reported here, of whom 40% were rituximab refractory and 20% refractory to last prior therapy, the GALEN regimen showed promising efficacy compared with a report of lenalidomide plus rituximab.9 We found here an ORR (CR) of 63.2% (57.9%) at the end of 6 cycles, and PFS, DOR, and OS of 52.1%, 68.4%, and 73.3%, respectively, after 3 years. The 52.1% 3-year PFS rate achieved with 6 cycles of lenalidomide and 8 infusions of obinutuzumab seems promising compared with the 52% 2-year time-to-progression rate seen in patients with relapsed but not refractory FL who received 12 cycles of lenalidomide but only 4 infusions of rituximab.9 Our data are consistent with the preliminary report of another phase 1 study that combined obinutuzumab and lenalidomide using a slightly different initial treatment schedule (no 8 days of lenalidomide prophase and a longer induction period of 12 months); no DLTs were observed at the highest dose of 20 mg, which was selected as the RP2D.22
Correlation of pharmacodynamic and biomarker studies is essential to decipher signaling mechanisms and ensure optimal efficacy with a manageable toxicity profile. Lenalidomide has been proposed to reduce CD20 expression on malignant B cells from chronic lymphocytic leukemia, suggesting a potential antagonism between lenalidomide and anti-CD20 therapy in chronic lymphocytic leukemia.23 To our knowledge, this finding has not been explored in B-cell NHL. Here, we show that there seems to be no pharmacodynamic antagonism between obinutuzumab and lenalidomide, because CD20 expression remained unaffected by lenalidomide (Figure 3). Functional experiments in samples from GALEN patients are under way to determine whether lenalidomide is able restore in vivo the capacity of circulating T cells to mount an efficient immune synapse with malignant B cells and whether NK cell–related ADCC and phagocytosis of opsonized lymphoma cells by macrophages are modulated by the combination.
In summary, oral lenalidomide plus obinutuzumab is well tolerated and effective in patients with R/R FL. The recommended phase 2 dose of lenalidomide was established at 20 mg based on the increased incidence of grade 3/4 neutropenia in cycle 2 at 25 mg. To further explore the optimal duration of this combination, the phase 2 part of this study is currently assessing efficacy of the GALEN regimen with a lenalidomide dose of 20 mg during 6 months and 2 years of maintenance (lenalidomide at a lower dose of 10 mg on days 2-22 of each cycle for a maximum of 12 cycles to mitigate hematologic toxicity, combined with obinutuzumab 1000 mg on day 1 every 2 cycles, then 1 additional year of obinutuzumab as a single agent every 2 cycles) in 3 separate populations of 90 to 100 patients: R/R aggressive lymphoma (cohort 1: diffuse large B-cell lymphoma and mantle cell lymphoma), R/R FL (cohort 2), and first-line FL with Groupe d’Etude des Lymphomes Folliculaires criteria (cohort 3).
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors acknowledge the editorial assistance provided by Laura Ninger and Kathleen Ohleth (Precise Publications, LLC). The authors thank the patients and their families, the Lymphoma Academic Research Organization team for the management of the study, the reviewers at the Lymphoma Study Association, and the independent data monitoring committee (André Bosly, Catherine Sebban, and Natacha Heutte).
This work was supported by investigator-initiated study grants from Celgene and Roche.
Authorship
Contribution: F.M. and R.H. designed the study; all authors participated in the collection and assembly of data; F.M., R.H., C.M., and K.T. provided data analysis and interpretation; F.M. was primarily involved in the manuscript writing; and all authors participated in the manuscript development process and provided final approval of the manuscript for submission.
Conflict-of-interest disclosure: F.M. has received personal fees for consultancy, advisory boards, or scientific lectures from Gilead, Janssen, Celgene, Servier, Bristol-Myers Squibb, Roche, and Epizyme (outside the submitted work); S.L.G. received research grants, personal fees, and nonfinancial support from Celgene and Roche Genentech during the conduct of the study and has received grants, personal fees, and nonfinancial support from Celgene outside the submitted work; H.T. has received research grants and personal fees from Celgene, personal fees and nonfinancial support from Roche, and personal fees from Karyopharm, AstraZeneca, and Bristol-Myers Squibb outside the submitted work; C.T. has received consultancy fees or honoraria from Celgene, Bayer, AbbVie, and Janssen and research funding from Roche; C.M. has received research funding from Celgene and honoraria from Celgene and Bristol-Myers Squibb; K.T. has received research funding from Celgene and honoraria from Celgene and Roche; G.C. has received personal fees for consultancy and honoraria from Roche and Celgene and received personal fees for honoraria from Sanofi, Gilead, and Janssen during the conduct of the study; and R.H. has received consulting fees or honoraria from Bristol-Myers Squibb and Gilead. The remaining authors declare no competing financial interests.
Correspondence: Franck Morschhauser, Université de Lille, CHU Lille, EA 7365–GRITA–Groupe de Recherche sur les formes Injectables et les Technologies Associées, F-59000 Lille, France; e-mail: franck.morschhauser@chru-lille.fr.
