

Abstract
Monoclonal gammopathy is a common condition, particularly in the elderly. It can indicate symptomatic multiple myeloma or another overt malignant lymphoid disorder requiring immediate chemotherapy. More frequently, it results from a small and/or quiescent secreting B-cell clone, is completely asymptomatic, and requires regular monitoring only, defining a monoclonal gammopathy of unknown significance (MGUS). Sometimes, although quiescent and not requiring any treatment per se, the clone is associated with potentially severe organ damage due to the toxicity of the monoclonal immunoglobulin or to other mechanisms. The latter situation is increasingly observed but still poorly recognized and frequently undertreated, although it often requires rapid specific intervention to preserve involved organ function. To improve early recognition and management of these small B-cell clone–related disorders, we propose to introduce the concept of monoclonal gammopathy of clinical significance (MGCS). This report identifies the spectrum of MGCSs that are classified according to mechanisms of tissue injury. It highlights the diversity of these disorders for which diagnosis and treatment are often challenging in clinical practice and require a multidisciplinary approach. Principles of management, including main diagnostic and therapeutic procedures, are also described. Importantly, efficient control of the underlying B-cell clone usually results in organ improvement. Currently, it relies mainly on chemotherapy and other anti–B-cell/plasma cell agents, which should aim at rapidly producing the best hematological response.
Introduction
Monoclonal gammopathy is defined by the presence in serum and/or urine of a monoclonal immunoglobulin produced by an abnormal B-cell clone. The clone usually consists of plasma cells when the monoclonal immunoglobulin (MIg) is immunoglobulin G (IgG), IgA, IgD, or light chain (LC) only and lymphoplasmacytic when producing an IgM. It may remain quiescent over a prolonged period, defining monoclonal gammopathy of undetermined significance (MGUS).1 Uncontrolled clonal expansion causing end-organ damage defines malignant symptomatic lymphoproliferative disorder, usually multiple myeloma (MM) or Waldenström macroglobulinemia (WM).2,3 An intermediate condition results in an indolent “smoldering” disease.4
Manifestations associated with monoclonal gammopathies can result from various, sometimes intertwined mechanisms: (1) expansion of clonal malignant cells; (2) secretion of a large amount of MIg, which can cause tumor-mass–related manifestations, including hyperviscosity and LC cast nephropathy; (3) secondary alterations of the host immune system causing immune deficiency and autoimmune manifestations; and (4) pathogenic activities of the MIg or other mediators released by the clone.
Quiescent or indolent B-cell clones do not cause tumor symptoms, and immunodepression is uncommon. However, even a very small clone can produce severe manifestations due to toxicity of the MIg or other mechanisms. The kidney is a frequent target and the concept of monoclonal gammopathy of renal significance (MGRS) has recently emerged.5 Other organs, particularly the skin and peripheral nerve, may be involved. To better characterize these situations caused by a “dangerous small B-cell clone,”6 which are still poorly recognized and frequently undertreated, we propose extending the concept of MGRS to that of monoclonal gammopathy of clinical significance (MGCS).
Types of MGCS according to mechanisms of tissue injury
The spectrum of MGCS is large because of the diversity of involved organs and pathogenic mechanisms. Lesions commonly result from deposition of all or part of the MIg as aggregates, amorphous, crystalline, microtubular, or fibrillar forms (Table 1). Other mechanisms include autoantibody activity against a tissue antigen, formation of immune complexes, and complement activation. In addition, even a small B-cell clone may absorb biologically active molecules or induce cytokine secretion. In some types of MGCS, the pathogenesis remains unknown (Table 2).
Current pathophysiological classification of the main MGCS-related disorders: MGCS due to deposition of all or part of the MIg
| Ultrastructural appearance of deposits | Main characteristics of monoclonal gammopathy | Main organ(s) involved | Reference | |
|---|---|---|---|---|
| Organized | ||||
| AL amyloidosis | Fibrillar | λ LC (75%), κ LC (25%), IgM <10% | Systemic (heart 80%, kidney 70%) | 8,57 |
| Type I cryoglobulinemia | Microtubular/crystalline | IgG or IgM | Systemic (skin +++, kidney, peripheral nerve, systemic symptoms [crystal cryoglobulin]) | 12,28 |
| Immunotactoid glomerulopathy/GOMMID | Microtubular | CLL-like clonal proliferation (50%) | Kidney | 13 |
| Acquired Fanconi syndrome | Crystalline | κ LC (>90%, mostly Vk1) | Kidney (proximal tubulopathy) | 9 |
| Crystal storing histiocytosis | Crystalline | κ LC | Systemic (kidney, cornea, joints, lymphoid tissue) | 10 |
| Crystalline keratopathy | Crystalline | IgG | Cornea | 11 |
| Nonorganized | ||||
| MIDD | LCDD: LC only (usually κ LC) (80%, Vk1 and Vk4) HCDD: truncated HC only (mostly γ1 and γ3) LHCDD: LC + truncated HC | Systemic (kidney [∼100%, glomerular and tubular basement membrane], liver [30%], heart [30%]) | 14,51,61 | |
| PGNMID | Usually IgG3 | Kidney | 15 | |
| Macroglobulinosis | IgM | Skin (dermis) | 16 |
| Ultrastructural appearance of deposits | Main characteristics of monoclonal gammopathy | Main organ(s) involved | Reference | |
|---|---|---|---|---|
| Organized | ||||
| AL amyloidosis | Fibrillar | λ LC (75%), κ LC (25%), IgM <10% | Systemic (heart 80%, kidney 70%) | 8,57 |
| Type I cryoglobulinemia | Microtubular/crystalline | IgG or IgM | Systemic (skin +++, kidney, peripheral nerve, systemic symptoms [crystal cryoglobulin]) | 12,28 |
| Immunotactoid glomerulopathy/GOMMID | Microtubular | CLL-like clonal proliferation (50%) | Kidney | 13 |
| Acquired Fanconi syndrome | Crystalline | κ LC (>90%, mostly Vk1) | Kidney (proximal tubulopathy) | 9 |
| Crystal storing histiocytosis | Crystalline | κ LC | Systemic (kidney, cornea, joints, lymphoid tissue) | 10 |
| Crystalline keratopathy | Crystalline | IgG | Cornea | 11 |
| Nonorganized | ||||
| MIDD | LCDD: LC only (usually κ LC) (80%, Vk1 and Vk4) HCDD: truncated HC only (mostly γ1 and γ3) LHCDD: LC + truncated HC | Systemic (kidney [∼100%, glomerular and tubular basement membrane], liver [30%], heart [30%]) | 14,51,61 | |
| PGNMID | Usually IgG3 | Kidney | 15 | |
| Macroglobulinosis | IgM | Skin (dermis) | 16 |
AL amyloidosis is the most frequent type, with an incidence of ∼10 per million inhabitants per year in Western countries. MIDD incidence is estimated to be 10-fold lower. Other types of MGCS due to deposition of MIg are rare (type I cryoglobulinemia and PGNMID), even scarcer (Fanconi syndrome), or exceptional (macroglobulinosis and crystalline keratopathy).
CLL, chronic lymphocytic leukemia, GOMMID, glomerulonephritis with organized microtubular monoclonal immunoglobulin deposits; HCDD, heavy-chain deposition disease; LCDD, light-chain deposition disease; LHCDD, light- and heavy-chain deposition disease.
Current pathophysiological classification of the main MGCS-related disorders: cytokine mediated or MGCS due to autoantibody activity, CAP activation, cytokine-mediated, or of unknown mechanism
| Mechanism | Main characteristics of monoclonal gammopathy | Main organ(s) involved | Reference | |
|---|---|---|---|---|
| Autoantibody activity | ||||
| Type II mixed cryoglobulinemia* | Rheumatoid | IgM | Immune complex–mediated vasculitis; skin +++, kidney, peripheral nerve; may be systemic | 28,60 |
| C1 inhibitor deficiency | C1 inhibitor | Angioedema | 25 | |
| Von Willebrand disease | vW factor | Bleeding | 26 | |
| Bullous skin diseases | Dermoepidermal junction (collagen VII) | Skin | 21 | |
| Xanthomatosis | Various lipoproteins | Usually IgG | Cholesterol accumulation in macrophages; skin and tendons; other localizations (necrobiotic xanthogranumathosis) | 29,30 |
| Cold agglutinin disease | Red blood cell (Ii) | IgM | Cold-induced skin manifestations + intravascular hemolysis | 27 |
| IgM-associated peripheral neuropathy | MAG +++ | IgM | Peripheral nerve; ataxic polyneuropathy (anti-MAG) CANOMAD | 22,23 |
| Gangliosides | ||||
| CAP*activation | ||||
| C3 glomerulonephritis Atypical hemolytic-uremic syndrome | Mechanism to be determined; autoantibody activity against CAP regulator protein (factor H) in some cases | IgG | Kidney only Systemic | 31,,-34 |
| Cytokine mediated | ||||
| POEMS syndrome | VEGF | λ LC (∼100%), | Peripheral nerve (100%) and various other manifestations | 35,36 |
| IgA 50% | ||||
| Vλ1 (#100%) | ||||
| Osteosclerotic bone lesions | ||||
| Unknown mechanism | ||||
| Systemic capillary leak syndrome | IgG, IgA (rare) | Systemic | 45 | |
| TEMPI syndrome | IgG | Systemic | 46,47 | |
| Neutrophilic dermatosis† | IgA >80% (except Sweet syndrome) | Skin +++; different types and different associated manifestations | 39 | |
| Acquired cutis laxa | Usually IgG; association with γ HCDD | Skin +++Other manifestations (lung, digestive tract) | 43 | |
| Scleromyxedema | IgG with slow electrophoretic mobility | Skin +++; other localizations | 40,41,52 | |
| Scleroedema | IgG | Skin only | 41 | |
| Schnitzler syndrome | Acquired autoinflammatory syndrome by IL-1 deregulation? | IgM | Skin +++; systemic symptoms; osteosclerotic bone lesions | 44 |
| Sporadic late-onset nemaline myopathy | Exclusively muscles (skeletal and possibly cardiac) | 48 |
| Mechanism | Main characteristics of monoclonal gammopathy | Main organ(s) involved | Reference | |
|---|---|---|---|---|
| Autoantibody activity | ||||
| Type II mixed cryoglobulinemia* | Rheumatoid | IgM | Immune complex–mediated vasculitis; skin +++, kidney, peripheral nerve; may be systemic | 28,60 |
| C1 inhibitor deficiency | C1 inhibitor | Angioedema | 25 | |
| Von Willebrand disease | vW factor | Bleeding | 26 | |
| Bullous skin diseases | Dermoepidermal junction (collagen VII) | Skin | 21 | |
| Xanthomatosis | Various lipoproteins | Usually IgG | Cholesterol accumulation in macrophages; skin and tendons; other localizations (necrobiotic xanthogranumathosis) | 29,30 |
| Cold agglutinin disease | Red blood cell (Ii) | IgM | Cold-induced skin manifestations + intravascular hemolysis | 27 |
| IgM-associated peripheral neuropathy | MAG +++ | IgM | Peripheral nerve; ataxic polyneuropathy (anti-MAG) CANOMAD | 22,23 |
| Gangliosides | ||||
| CAP*activation | ||||
| C3 glomerulonephritis Atypical hemolytic-uremic syndrome | Mechanism to be determined; autoantibody activity against CAP regulator protein (factor H) in some cases | IgG | Kidney only Systemic | 31,,-34 |
| Cytokine mediated | ||||
| POEMS syndrome | VEGF | λ LC (∼100%), | Peripheral nerve (100%) and various other manifestations | 35,36 |
| IgA 50% | ||||
| Vλ1 (#100%) | ||||
| Osteosclerotic bone lesions | ||||
| Unknown mechanism | ||||
| Systemic capillary leak syndrome | IgG, IgA (rare) | Systemic | 45 | |
| TEMPI syndrome | IgG | Systemic | 46,47 | |
| Neutrophilic dermatosis† | IgA >80% (except Sweet syndrome) | Skin +++; different types and different associated manifestations | 39 | |
| Acquired cutis laxa | Usually IgG; association with γ HCDD | Skin +++Other manifestations (lung, digestive tract) | 43 | |
| Scleromyxedema | IgG with slow electrophoretic mobility | Skin +++; other localizations | 40,41,52 | |
| Scleroedema | IgG | Skin only | 41 | |
| Schnitzler syndrome | Acquired autoinflammatory syndrome by IL-1 deregulation? | IgM | Skin +++; systemic symptoms; osteosclerotic bone lesions | 44 |
| Sporadic late-onset nemaline myopathy | Exclusively muscles (skeletal and possibly cardiac) | 48 |
CAP, complement alternative pathway; CANOMAD, chronic ataxic neuropathy, ophthalmoplegia, monoclonal IgM protein, cold agglutinins, anti-disialosyl antibodies; HCDD, heavy-chain deposition disease; IL-1, interleukin 1; POEMS, polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy, and skin changes; TEMPI, telangiectasias, erythrocytosis with elevated erythropoietin level, monoclonal gammopathy, perinephric fluid collection, and intrapulmonary shunting.
Type II mixed cryoglobulinemia was observed in ∼10% of patients with chronic virus C infection.
Including pyoderma gangrenosum, Sweet syndrome, subcorneal pustular dermatosis, and erythema elevatum diutinum.
MGCS due to MIg deposition
MIg deposition is involved in many MGCS types, which may be classified according to the ultrastructural appearance of deposits, either organized or nonorganized (Table 1).7
Organized deposits display different patterns. Amyloid light-chain (AL) amyloidosis is the hallmark of MGCS with fibrillar deposits.8 Acquired Fanconi syndrome features precipitation of LC (usually κ) into crystals within renal proximal tubular cells.9 Accumulation of MIg crystals in the lysosomes of macrophages within bone marrow, lymph nodes, and extralymphoid tissue characterizes crystal storing histiocytosis.10 Crystalline keratopathy usually results from formation of monoclonal immunoglobulin-derived crystals in the cornea.11 Some type I cryoglobulins are crystalglobulins. Monoclonal and mixed cryoglobulins may display microtubular deposits with or without vasculitic lesions, depending on cryoglobulin composition and involved organ.7,12 Immunotactoid glomerulopathy or glomerulonephritis with organized microtubular MIg deposits is due to microtubular IgG deposits (usually limited to the kidney), without features of cryoglobulinemic glomerulonephritis.7,13
MGCS with nonorganized deposits are mainly represented by monoclonal immunoglobulin deposition disease (MIDD), featured by linear granular “powdery punctate” deposits along basal membranes. Usually, deposits contain the LC (LCDD), sometimes the heavy chain (HCDD), or both (LHCDD). Although predominantly involving glomerular and tubular compartments of the kidney, MIDDs are systemic.7,14 In proliferative glomerulonephritis with monoclonal immunoglobulin deposits (PGNMID), the MIg (most commonly IgG3) and complement components are deposited in the glomerular capillary walls and mesangium, mimicking an immune complex–mediated glomerulonephritis7,15 (Figure 1A-C). In contrast to MIDD, PGNMID is limited to the kidney. Uncommonly, amorphous MIg deposits may involve other tissues, as in macroglobulinosis, featured by intradermis deposits of monoclonal IgM.16 Glomerular intracapillary monoclonal IgM thrombi, a now rare complication of WM with hyperviscosity,17 and LC cast nephropathy, which results from massive nephrotoxic LC secretion, indicate a high tumor burden and do not belong to the spectrum of MGCS.18

Histopathological features of MGCS-related PGNMID and C3 glomerulonephritis. PGNMID (A-C). (A) Light microscopy with periodic acid–Schiff staining (original magnification ×400; scale bar, 50 μm). Membranoproliferative glomerulonephritis with mesangial and endocapillary hypercellularity. (B) Immunofluorescence study showing monotypic IgG glomerular deposits that stained positive with the anti-gamma3 conjugate. Similar staining was observed with the anti-κ conjugate, whereas staining with conjugates specific for α, μ, gamma1, gamma2, and gamma4 heavy chains and λ light-chain conjugate was negative (not depicted). Scale bar, 50 µm. (C) Electron microscopy confirmed the diagnosis of PGNMID with granular nonorganized electron-dense deposits predominant in the subendothelial space (asterisk), suggestive of immune complex–mediated glomerulonephritis (original magnification ×15 000; scale bar, 1 µm). (D-F) MGCS-associated C3 glomerulonephritis. (D) Light microscopy showing a pattern of proliferative endocapillary glomerulonephritis (periodic acid-Schiff staining; original magnification ×400; scale bar, 50 μm). (E) By immunofluorescence, mesangial and glomerular capillary wall deposits stained positively with the anti-C3 FITC-conjugate (original magnification ×200), whereas no staining was observed with conjugates specific for γ, α, and μ heavy chains and with anti-κ and anti-λ conjugates (not depicted). Scale bar, 50 µm. (F) Electron microscopy demonstrated the presence of voluminous subepithelial electron-dense deposits (humps, asterisk) and interrupted intramembranous dense deposits (arrows) (original magnification ×15 000; scale bar, 1 µm)
Histopathological features of MGCS-related PGNMID and C3 glomerulonephritis. PGNMID (A-C). (A) Light microscopy with periodic acid–Schiff staining (original magnification ×400; scale bar, 50 μm). Membranoproliferative glomerulonephritis with mesangial and endocapillary hypercellularity. (B) Immunofluorescence study showing monotypic IgG glomerular deposits that stained positive with the anti-gamma3 conjugate. Similar staining was observed with the anti-κ conjugate, whereas staining with conjugates specific for α, μ, gamma1, gamma2, and gamma4 heavy chains and λ light-chain conjugate was negative (not depicted). Scale bar, 50 µm. (C) Electron microscopy confirmed the diagnosis of PGNMID with granular nonorganized electron-dense deposits predominant in the subendothelial space (asterisk), suggestive of immune complex–mediated glomerulonephritis (original magnification ×15 000; scale bar, 1 µm). (D-F) MGCS-associated C3 glomerulonephritis. (D) Light microscopy showing a pattern of proliferative endocapillary glomerulonephritis (periodic acid-Schiff staining; original magnification ×400; scale bar, 50 μm). (E) By immunofluorescence, mesangial and glomerular capillary wall deposits stained positively with the anti-C3 FITC-conjugate (original magnification ×200), whereas no staining was observed with conjugates specific for γ, α, and μ heavy chains and with anti-κ and anti-λ conjugates (not depicted). Scale bar, 50 µm. (F) Electron microscopy demonstrated the presence of voluminous subepithelial electron-dense deposits (humps, asterisk) and interrupted intramembranous dense deposits (arrows) (original magnification ×15 000; scale bar, 1 µm)
MGCS due to autoantibody activity of MIg
MIg can cause organ lesions through autoantibody activity against various antigens. Anti-collagen IV or anti-phospholipase A2 receptor reactivity of the MIg was documented in rare cases of antiglomerular basement membrane disease and membranous glomerulopathy, respectively.19,20 MIg-related bullous skin diseases were reported due to autoantibody activity versus collagen VII.21 The diagnosis requires pathological demonstration of linear immunoglobulin deposits in the dermoepidermal junction with the same LC restriction as that of the circulating MIg. Alternatively, immunostaining may show polytypic deposits, indicating that the autoantibody activity is not MIg related but produced by bystander autoreactive polyclonal B cells, as in autoimmune cytopenias associated with lymphoid disorders.21
An autoantibody activity against myelin-associated glycoprotein (MAG) may be responsible for IgM-associated peripheral neuropathy.22 The MIg usually recognizes a myelin-sulfated glucuronyl epitope, shared by MAG and cross-reactive glycoconjugates. Predominantly sensitive symmetric and distal demyelinating polyneuropathy often accompanied by ataxia and tremor is the main clinical manifestation. Monoclonal IgM autoantibodies against gangliosides containing disialosyl groups are less frequent and usually observed in CANOMAD syndrome (chronic ataxic neuropathy, ophthalmoplegia, monoclonal IgM protein, cold agglutinins, anti-disialosyl antibodies).22,23 Antibodies against GM1 ganglioside associated with multifocal motor neuropathy are usually of IgM class but polytypic; whether monoclonal IgM with anti-GM1 specificity can cause weakness and motor conduction blocks is debated.24
Acquired C1 inhibitor deficiency and acquired von Willebrand disease can be due to autoantibody activity of MIg with direct inhibitory effect or inducing formation of complexes accelerating the catabolism of the protein.25,26
In cold agglutinin disease, cold-triggered ischemic symptoms of the extremities and acute hemolysis are provoked by agglutination of red blood cells due to the anti-Ii autoantibody activity of the monoclonal IgM.27 In type II mixed cryogloglobulinemia, monoclonal IgM with rheumatoid activity triggers the deposition of immune complexes with polyclonal IgG in vascular walls, resulting in complement activation and formation of perivascular infiltrates characteristic of leukocytoclastic vasculitis.28
MIg autoantibodies can become symptomatic by intracellular accumulation of immune complexes. Xanthomatosis is characterized by the intracellular storage of cholesterol-rich material in macrophages, accumulating in the skin and the tendons29 (Figure 2A). Xanthomas may be diffuse, predominating in the limb skinfolds, or associated with an inflammatory reaction referred to as necrobiotic xanthogranulomatosis.30 Particularly in the normolipemic form, xanthomatosis is associated with MIg and complement abnormalities with low C4 serum levels. The MIg may be involved through antigen-antibody interaction with various lipoproteins resulting in the formation of immune complexes, complement activation, and subsequent lipid accumulation in macrophages.29
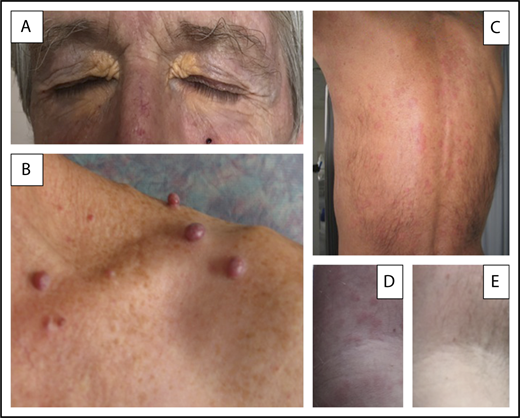
Three different MGCS-associated skin manifestations. (A) Normolipemic xanthoma with marked periorbital involvement in a patient with decreased total complement activity (CH50) and serum C4 level suggesting classical complement pathway activation by immune complexes. (B) Glomeruloid hemangiomata in a patient with POEMS syndrome, likely resulting from the effects of VEGF on angiogenesis. (C-E) Skin lesions of Schnitzler syndrome. (C) Typical pseudourticarial aspect predominantly involving the back. (D-E) Close-up view of lesions of the upper limb in another patient illustrating the efficacy of anakinra and showing initial lesions (D) and normal aspects (E) of the skin obtained after the first injection of the IL-1 antagonist. Skin lesions did not recur after 2 years of follow-up from diagnosis on continuously maintained daily treatment.
Three different MGCS-associated skin manifestations. (A) Normolipemic xanthoma with marked periorbital involvement in a patient with decreased total complement activity (CH50) and serum C4 level suggesting classical complement pathway activation by immune complexes. (B) Glomeruloid hemangiomata in a patient with POEMS syndrome, likely resulting from the effects of VEGF on angiogenesis. (C-E) Skin lesions of Schnitzler syndrome. (C) Typical pseudourticarial aspect predominantly involving the back. (D-E) Close-up view of lesions of the upper limb in another patient illustrating the efficacy of anakinra and showing initial lesions (D) and normal aspects (E) of the skin obtained after the first injection of the IL-1 antagonist. Skin lesions did not recur after 2 years of follow-up from diagnosis on continuously maintained daily treatment.
MGCS secondary to CAP activation
In patients >50 years of age, C3 glomerulopathy (C3G) is frequently associated with an indolent IgG monoclonal gammopathy.31,32 C3G, which is defined histologically by the presence of glomerular electron dense deposits of C3 without immunoglobulin deposit, derives from activation of the CAP (Figure 1D-F). The associated MIg probably plays a role, as suggested by the demonstration in some cases of its autoantibody activity against CAP regulatory proteins, such as factor H, and by the beneficial effect of a clone-targeted chemotherapy.32 In atypical hemolytic uremic syndrome, the prevalence of monoclonal gammopathy in older adults is also increased and an autoantibody activity against factor H of the MIg has been similarly reported.33,34 Altogether, these data suggest an under-recognized interaction between monoclonal gammopathy and CAP regulation that deserves further investigation. It may involve several mechanisms, since an anticomplement autoantibody activity of the MIg appears uncommon.31,32
MGCS due to cytokine secretion
The acronym POEMS refers to the association of a plasma cell disorder with polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy, and skin changes35 (Figure 2B). The MIg is almost always of the λ type with an overrepresentation of IgA. The bone marrow usually contains <5% plasma cells. Bone lesions, most commonly osteosclerotic, are almost always present and are usually focal. Their detection requires a careful workup. Indeed, efficient treatment of the plasma cell disorder, such as the irradiation of a solitary plasmocytoma, usually results in resolution of all manifestations. A characteristic feature is the marked elevation of serum vascular endothelial growth factor (VEGF) levels that correlates with disease activity.35 VEGF action on vascular permeability and angiogenesis could account for edema in the myelin sheath, causing peripheral nerve involvement and skin manifestations, such as angiomata. In addition, osteosclerotic bone lesions could result from VEGF-induced osteoblastic differentiation. Thus, abnormal VEGF secretion appears to play a key pathogenic role. The λ type restriction of the MIg and molecular studies showing that secreting clonal cells use only 2 Vλ1 germ line genes36 suggest an interaction with VEGF production through potential autoantibody activity or a ligand-receptor effect.
MGCS of unknown or putative mechanism
Monoclonal gammopathy may cause various manifestations (particularly bleeding disorders) related to adsorption of biologically active molecules on clonal cells or aggregated MIg. This is exemplified by GpIb-mediated selective adsorption of von Willebrand factor on clonal plasma cells37 and by factor X adsorption on AL amyloid fibrils.38 MIg-mediated impaired platelet function is a classical complication of symptomatic lymphoproliferation with high MIg serum levels. Whether low MIg levels may affect platelet aggregation remains unclear.
In several diseases associated with a monoclonal gammopathy, the pathogenic link is likely but poorly understood. Most involve the skin, including the group of neutrophilic dermatosis that encompasses pyoderma gangrenosum, Sweet syndrome, subcorneal pustular dermatosis, and erythema elevatum diutinum. Except for Sweet syndrome, >80% of cases of neutrophilic dermatosis with MIg are featured by monoclonal IgA, suggesting a role of IgA, IgA receptor, and/or mucosal immunity in the emergence of neutrophilic tissular infiltrations.39
The presence of a monoclonal IgG is a diagnostic criterion of scleromyxedema, characterized by papular and sclerodermoid skin lesions due to dermal mucin deposition, fibroblast proliferation, and fibrosis, with potential systemic extension.40,41 Scleredema also featuring mucin deposition is restricted to the skin and mainly affects the back and shoulders.41 A monoclonal gammopathy is also frequent in acquired cutis laxa that manifests with the decreased elasticity of the skin and other tissues due to the immunoglobulin-mediated breakdown of elastic fibers. In some patients with γ-HCDD, dermal deposition of monoclonal γ heavy chains may result in elastic tissue destruction through complement-mediated release of elastases.42,43 Schnitzler syndrome is defined by the combination of a monoclonal IgM with recurrent urticarial rash and mild dermal perivascular neutrophilic infiltrates, without significant immunoglobulin or complement deposition.44 The IL-1 receptor antagonist anakinra produces a dramatic improvement of all manifestations, including fever, fatigue, musculoskeletal pain, and biological markers of inflammation, which frequently accompany skin symptoms (Figure 2C-E). Accordingly, Schnitzler syndrome may represent an acquired counterpart of hereditary autoinflammatory disorders also characterized by skin rashes and recurrent fever, in which IL-1 secretion arises from deregulation of the inflammasome.44 Most patients with idiopathic systemic capillary leak syndrome (Clarkson disease) have a detectable serum MIg, whose role in increased capillary permeability and subsequent loss of protein-rich fluid to the interstitial space is unclear.45 A monoclonal gammopathy (usually an IgGk) is often associated with TEMPI syndrome, which includes telangiectasias, erythrocytosis with elevated erythropoietin levels, perinephric-fluid collections, and intrapulmonary shunting.46 The efficacy of the proteasome inhibitor bortezomib supports a causal role of the gammopathy.47 Similar therapeutic data suggest a relationship between sporadic late onset nemaline myopathy and MGUS.48
Diagnostic considerations
The diagnosis of MGCS implicates excluding a chance association because of the high prevalence of MIg, particularly in the elderly.49 Establishing a causal relationship between monoclonal gammopathy and clinical manifestations may depend on the pathological demonstration of MIg deposition in the affected tissue or organ. Immunohistological techniques using antibodies specific for LC isotypes and, when appropriate, IgG subclasses should be used to demonstrate that immunoglobulin deposits match the circulating MIg. Pathological studies may require electron microscopy, which allows for proper characterization of the ultrastructural organization of immunoglobulin deposits.7 Importantly, the diagnosis of AL amyloidosis requires not only Congo red staining of a biopsied tissue but the demonstration of the immunoglobulinic nature of the fibrils using immunohistochemistry and, eventually, immunoelectron microscopy or mass spectrometry.8 This is necessary to exclude the coincidental association of a monoclonal gammopathy with a late-onset hereditary form of amyloidosis or the wild-type transthyretin (senile cardiac amyloidosis). Indeed, approximately one-quarter of patients with senile amyloidosis (usually elderly males) present with a MIg.50
For the diagnosis of a MIg-mediated immune process, a high titer of autoantibody activity is important. The specificity of the MIg should be defined and correlated with clinical data. In IgM-mediated neuropathies, sensitive immunoblotting or enzyme-linked immunosorbent assays are usually required, since the monoclonal IgM may target hidden epitopes of nerve glycolipids or glycoproteins.22,23 Measurement of serum complement levels is often useful. Low CH50 and C4 levels suggest activation of the complement classical pathway by immune complexes, as in type II cryoglobulinemia and xanthomatosis, or by MIg aggregates, as in type I cryoglobulinemia. In γ-HCDD, activation of the classical pathway is also common, favored by partial deletion of the constant domain.51 Low C3 levels suggesting CAP activation may be observed in C3G and thrombotic microangiopathy associated with MIg.32
For conditions of unknown mechanism, the link with the associated MIg is primarily suggested by epidemiological data. In Schnitzler syndrome and scleromyxedema, the association is so frequent that the presence of MIg contributes to defining the disease.40,44 Additionally, in scleromyxedema, the monoclonal IgG shows characteristic slow electrophoretic mobility.52 In all situations where the pathogenic role of the MIg is questionable, comparison of V gene usage may be helpful for documenting common antibody reactivity.36 Even if epidemiologic and immunological data suggest a relationship, the pathogenic role of the MIg remains putative, since the secreting B-cell clone may have developed secondarily to the associated condition. Thus, in primary hyperparathyroidism, an association with monoclonal gammopathy has been reported and the emergence of the plasma cell clone in response to parathormone-induced IL-6 secretion has been hypothesized.53 Finally, clinical evidence may corroborate a causal link when therapeutic measures targeting the MIg or the secreting B-cell clone are effective, as in C3G,32 TEMPI syndrome,47 nemaline myopathy,48 and scleromyxedema.54
Treatment considerations
Indication for therapy in MGCS is driven by organ damage due to the secreting clone, even though it would not have required any therapy per se. Currently, targeting this small “dangerous” B-cell clone relies upon anti–B-cell/plasma cell agents, including monoclonal antibody and, in selected cases, local irradiation.5
Treatment decisions should be made using a benefit-to-risk approach while considering involved organs and natural disease history. No one would question giving polychemotherapy or even high-dose therapy with autotransplantation to a patient with MGCS and AL amyloidosis. In contrast, these therapeutic options are rarely used in patients with MGCS and a slowly progressive skin disorder.
Although treatment strategy should aim at producing the best hematological response, it should be also tailored to patient’s risk factors, as illustrated by AL amyloidosis.8 If the clone is plasmacytic, secreting a monoclonal IgG, IgA, or LC, then treatment should be based on anti-MM agents.56 The current strategy mainly relies on repeated courses of bortezomib, usually combined with an alkylating agent and dexamethasone (eg, the CyborD regimen) that produces rapid and deep hematological response. In addition, bortezomib does not require dose adaptation in patients with impaired renal function.55 In case of relapsing or refractory disease, immunomodulatory drugs should be considered. Anti-CD38 monoclonal antibodies, which are highly effective in symptomatic MM and currently being evaluated in AL amyloidosis,57 are likely to be increasingly used in the treatment of various MGCS.
If the clone is lymphoplasmacytic or corresponds to chronic lymphocytic leukemia or B-cell lymphoma, then treatment must be adapted accordingly, usually based on an anti-CD20 monoclonal antibody. When lymphoplasmacytic and associated with an IgM, the clone should be treated similarly to WM.58 The place of Bruton tyrosine kinase inhibitors and other nonconventional new agents remains to be assessed.58,59 Of note, whereas an anti-CD20 monoclonal antibody is appropriate in IgM-related disorders, particularly symptomatic type II cryoglobulinemia,60 it is not indicated for treating a plasmacytic clone, whose cells are usually CD20 negative.
Treatment management relies on the assessment of hematological response by serial measurement of the pathogenic MIg, most commonly its LC.55 Repeat evaluation of involved serum free LC is crucial in AL amyloidosis and LCDD, since the quality of the hematological response conditions organ response and patient survival.8,61 Why this correlation is less clear in anti-MAG IgM-associated polyneuropathy remains to be determined.22 However, achievement of the best hematological response is the goal of treatment and the main criterion for determining its optimal duration.
Besides targeting the pathogenic clone, other strategies can be considered, depending on each MGCS type. In MGCS due to tissue deposition, particularly AL amyloidosis, potential clearance of already deposited MIg material is currently under investigation based on therapeutic monoclonal antibodies against amyloid conformational neoepitope or serum amyloid P component
High-dose IV immunoglobulins are sometimes proposed as an alternative to chemotherapy, although their efficacy is nearly always temporary. Long-term treatment may maintain benefit, but availability, cost, and side effects (including renal toxicity) are concerns. Mechanisms of action may involve the inhibition of autoantibody activity, complement deviation, and cellular response.62 IV immunoglobulins may be effective in some MGCS of autoimmune origin, such as anti-ganglioside IgM-associated polyneuropathy, but not in others (including anti-MAG polyneuropathy).23 Systemic capillary leak syndrome63 or scleromyxedema40 may be improved, arguing in favor of an autoimmune activity of the MIg.
Conclusion
The evaluation of any MIg discovered by chance should be based on a clinical approach, including careful clinical examination and analysis of proteinuria. Conversely, searching for a MIg is justified in the presence of a wide range of manifestations. These approaches may lead to the identification of a small dangerous B-cell clone requiring aggressive therapy because of its potentially devastating consequences. To clearly distinguish this clonal disorder from an asymptomatic indolent gammopathy “of undetermined significance,” we introduce the concept of MGCS, thus extending the already established concept of MGRS. Relevant situations should be recognized by combining the term MGCS with disease characterization, introducing, for instance, the terminology “AL amyloidosis-related MGCS” or “cryoglobulinemia-related MGCS” to separate these conditions from “MM with AL amyloidosis” or “WM with cryoglobulinemia.” Because of its therapeutic implications, MGCS must be considered as a separate entity within the spectrum of monoclonal gammopathies.
Authorship
Contribution: All authors participated in the conception and in the writing of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Jean-Paul Fermand, Division of Immuno-Hematology, Hôpital Saint-Louis, 1 Ave Claude Vellefaux, 75010 Paris, France; e-mail: jpfermand@yahoo.fr.
