Key Points
IV therapy with oncolytic VSV-IFNβ-NIS virus extends survival of immunocompetent mice with AML.
The therapeutic outcome of VSV-IFNβ-NIS–treated mice with AML is augmented by anti-PD-L1 immunotherapy.
Abstract
Patients with relapsed acute myeloid leukemia (AML) have limited therapeutic options. Vesicular stomatitis virus (VSV)–interferon β (IFNβ)–sodium iodide symporter (NIS) is an oncolytic VSV encoding IFNβ and the NIS reporter. Syngeneic AML C1498 tumors responded to IV therapy with VSV-murine IFNβ (mIFNβ)-NIS in a dose-dependent manner. Imaging for NIS expression showed robust virus infection within the tumors. Virus infection did not increase programmed death ligand 1 (PD-L1) on tumor cells. Combining VSV-mIFNβ-NIS with anti-PD-L1 antibody (Ab) therapy enhanced antitumor activity compared with treatment with virus alone or Ab alone; this enhancement was not significant at higher VSV-mIFNβ-NIS doses. Systemic VSV therapy reduced systemic C1498–green fluorescent protein (GFP) tumor burden in the blood, bone marrow, spleen, and liver of mice with AML. Combination VSV-mIFNβ-NIS and anti-PD-L1 Ab therapy significantly enhanced the survival of these mice with no evidence of toxicity, compared with isotype control, anti-PD-L1, or virus alone. There was an increase in tumor-infiltrating CD4 and CD8 cells. Single-agent VSV-mIFNβ-NIS virotherapy induced both VSV-specific and GFP-specific CD8 T cells as determined by IFN-γ enzyme-linked immunospot, pentamer, and intracellular IFN-γ staining assays. Both of these responses were further enhanced by addition of anti-PD-L1 Ab. Depletion of CD8 or natural killer cells, but not CD4 cells, resulted in loss of antitumor activity in the VSV/anti-PD-L1 group. Clinical samples from chronic myelomonocytic leukemia and acute myelomonocytic leukemia appear to be especially susceptible to VSV. Overall, our studies show that oncolytic virotherapy combined with immune checkpoint blockade is a promising approach to AML therapy.
Introduction
Acute myeloid leukemia (AML) is a clonal hematopoietic stem cell disorder, usually characterized by ≥20% bone marrow blasts and is associated with significant mortality and morbidity.1 Chemotherapeutic regimens combining anthracyclines and cytarabine have served as the treatment backbone for this disease for several years, providing complete remission rates of 60% to 80% in young adults.2-4 Although therapeutic advances may have benefited specific AML subtypes (eg, Fms-like tyrosine kinase 3 [FLT3] inhibitors in FLT3-mutated AML), in general, newer agents have not added much to the induction of remission rates.3 In the majority of patients with AML, unless intense consolidative strategies such as allogeneic stem cell transplant are undertaken, the relapse rates are high. In general, the prognosis of patients after relapse is poor and treatment options are often unsatisfactory.
Recent preclinical and clinical data have emphatically demonstrated the activity of live replication-competent tumor-selective (oncolytic) viruses against hematologic malignancies.5-7 The most notable example is the first clinical demonstration of complete remission of disseminated plasmacytomas and clearance of myeloma cells in the bone marrow of a patient treated with a high IV dose of oncolytic measles virus encoding the sodium iodide symporter (NIS) as a reporter gene.8 Using longitudinal NIS and 123I/single-photon emission computed tomography (SPECT)/computed tomography (CT) imaging, the authors demonstrated selective infection of the disseminated tumors at days 8 and 15, followed by clearance of the virus infection by day 28. Another virus in clinical testing against myeloma is Coxsackievirus A21 (CVA21), an enterovirus that exhibited a potent cytostatic and cytocidal effect against myeloma.9 Reovirus, a positive strand RNA virus, has shown preclinical activity against non-Hodgkin lymphoma, chronic lymphocytic leukemia, and myeloma.10 Myxoma virus has antitumor activity against AML and myeloma tumor xenografts, and is able to target primary human leukemia cells while sparing normal hematopoietic stem and progenitor cells.11,12 We recently demonstrated that the Indiana strain of vesicular stomatitis virus (VSV), a negative-strand RNA virus, has potent oncolytic activity against human and murine myeloma tumors.13-15 VSV-interferon β (IFNβ)-NIS encodes human IFNβ (hIFNβ) or murine IFNβ (mIFNβ) to enhance tumor cell selectivity; it encodes the NIS transgene to facilitate noninvasive imaging of virus spread and enhance therapeutic efficacy with concurrent radioiodine therapy.13 Both VSV-hIFNβ-NIS and VSV-mIFNβ-NIS induced complete remission of 5TGM1 myeloma tumors in immunocompetent mice after systemic administration. Interestingly, tumor relapse rates were higher in mice treated with VSV encoding hIFNβ (biologically inactive in mice), supporting previous studies showing that IFNβ has a role in enhancing cross-priming and potentiating antitumor T-cell responses.16,17
Programmed death ligand 1 (PD-L1) is upregulated in many cancers and inhibits cytotoxic T-cell activity by binding to the programmed death 1 (PD-1) receptor on T cells. Blocking PD-L1 should therefore enhance anticancer immunity, and numerous anti-PD-L1 inhibitors are being tested in clinical trials. Predictive response markers are of interest; responses were observed in patients with tumors expressing high levels of PD-L1, especially when PD-1 was expressed by tumor-infiltrating immune cells.18,19 PD-L1 is upregulated in CD34+ cells from AML and chronic myelomonocytic leukemia (CMML) patients.18,20 In this study, we evaluated combining VSV virotherapy with anti-PD-L1 antibody (Ab) therapy in the C1498 murine AML model. This model was chosen because VSV replicated well in C1498 cells and killed the majority of cells within 48 hours in vitro. Of particular concern is the timely application of Ab to prevent premature immune clearance of virally infected cells. Hence, we also performed noninvasive imaging studies on a cohort of animals to monitor VSV-mIFNβ-NIS spread in the tumors, with and without anti-PD-L1 Ab treatment. Using a subcutaneous and a disseminated model of murine AML, we showed that coupling VSV virotherapy with immune checkpoint inhibitors could be a promising multimodal approach to improve the dismal prognosis for AML patients.
Materials and methods
Cells and viruses
The murine AML cell line C1498 (ATCC TIB-49) was transduced with a lentivirus encoding enhanced green fluorescent protein (GFP) and puromycin for selection (puromycin, 5 μg/mL) of GFP-positive cells (Imanis Life Sciences). Peripheral blood mononuclear cells (MNCs) and bone marrow (MNC) aspirates from AML patients were obtained after approval by the Mayo Clinic Institutional Review Board (IRB #14-006487) and with patient informed consent. The Indiana strain of recombinant VSV encoding GFP or mIFNβ and NIS has been described elsewhere.13,15 The recombinant viruses were propagated as previously described.21
Infectivity assays
Cells in 96-well plates were exposed to VSV-GFP or VSV-mIFNβ-NIS at multiplicity of infections of 0, 0.1, 1, and 10. Cell viability was assessed at 72 hours postinfection using the MTS cell proliferation assay (Promega). For infectious virus recovery assays, tumors were weighed and homogenized in 3 volumes (wt/vol) of Opti-MEM buffer. The clarified supernatant was titered on Vero cells using 50% tissue culture infective dose (TCID50) titrations.22
In vivo experiments
Animal protocols were approved by the Mayo Clinic Institutional Care and Use Committee. Four- to 5-week-old female C57BL/6J mice (Harlan Laboratories) received 2 × 106 C1498 cells implanted subcutaneously on the right flank. Fourteen days later, mice received 1 IV dose of VSV-mIFNβ-NIS (106, 107, or 108 TCID50) or phosphate-buffered saline. Three days later, some mice received 200 μg of anti-PD-L1 Ab (10F.9G2; BioXCell) given intraperitoneally (IP) on days 17, 20, and 23. Isotype control rat immunoglobulin G2b (IgG2b) Ab (LTF-2; BioXCell) was used in the control group. Tumors were harvested on day 30 (16 days postvirus).
AML was established by IV injection of 2 × 106 C1498.GFP cells into mice. Twelve days post-C1498.GFP administration, mice were treated with VSV-mIFNβ-NIS (108 TCID50; IV). Three days later, some mice received anti-PD-L1 Ab IP (200 μg per dose) 3 times, given 3 days apart. Rat IgG2b Ab was used as the isotype control. Mice were euthanized when they lost ≥10% of their body weight, demonstrated inactivity, or exhibited paralysis of both hindlimbs. T-cell depletion studies were performed to determine the impact of CD4, CD8, or natural killer (NK) cells on virotherapy. Details of the experiment and the associated immune cell analysis (enzyme-linked immunospot [ELISPOT] and IFN-γ staining) are in supplemental Methods (available on the Blood Web site).
SPECT/CT imaging
For imaging studies, mice received 1 IV administration of 300 μCi of 99mTc pertechnetate and were imaged 1 hour later on a micro-SPECT/CT scanner (MI Labs). DICOM data files were analyzed by Imanis Life Sciences to generate a normalized data set for comparison of virus intratumor distribution between treatment groups.
Statistical analysis
Statistical analysis of data was performed using GraphPad Prism (version 6.0a; GraphPad Software) using the unpaired Student t test; Kaplan-Meier survival curves were compared by log-rank (Mantel-Cox) analysis. A value of P < .05 was considered statistically significant.
Results
VSV-mIFNβ-NIS is active against subcutaneous AML tumors in a dose-dependent manner
C1498 is a murine AML cell line that grows as subcutaneous tumors or, if administered IV, establishes in the bone marrow, spleen, liver, and blood of syngeneic C57/B6 mice.23 In vitro, the cells are highly susceptible to infection and killing by VSV encoding GFP (VSV-GFP) or VSV-mIFNβ-NIS (supplemental Figure 1). To evaluate susceptibility of C1498 tumors to VSV infection and spread, C57/B6 mice with subcutaneous C1498 tumors were given VSV-mIFNβ-NIS (108 TCID50) IV; 3 days later, they were given 99mTc pertechnetate and imaged on a micro-SPECT/CT scanner. Isotope uptake was seen in the tumors of mice given VSV-mIFNβ-NIS but not in saline controls (Figure 1A). NIS-expressing infectious foci are clearly discernable from the high-resolution SPECT imaging data. It is apparent that the distribution of virus infection is not homogenously distributed in these C1498 tumors.

VSV therapy in subcutaneous C1498 tumors. (A) Representative SPECT/CT images of tumors (circled) in mice (n = 3 per group) that received VSV-mIFNβ-NIS, with and without anti-PD-L1 Ab (given 6 hours later). Eight serial sections through similar locations of the same mouse reveal NIS expression in VSV infectious foci distributed throughout the tumor. (B) Dose-dependent study on antitumor activity of VSV-mIFNβ-NIS against subcutaneous C1498 tumors. Mice were harvested 30 days post-tumor cell implantation and tumors were weighed. Mean ± SEM (n = 5 mice) from each group is plotted. *P ≤ .05. Unpaired Student t test was used. SEM, standard error of the mean.
VSV therapy in subcutaneous C1498 tumors. (A) Representative SPECT/CT images of tumors (circled) in mice (n = 3 per group) that received VSV-mIFNβ-NIS, with and without anti-PD-L1 Ab (given 6 hours later). Eight serial sections through similar locations of the same mouse reveal NIS expression in VSV infectious foci distributed throughout the tumor. (B) Dose-dependent study on antitumor activity of VSV-mIFNβ-NIS against subcutaneous C1498 tumors. Mice were harvested 30 days post-tumor cell implantation and tumors were weighed. Mean ± SEM (n = 5 mice) from each group is plotted. *P ≤ .05. Unpaired Student t test was used. SEM, standard error of the mean.
Mice with established subcutaneous tumors received 106, 107, or 108 TCID50 viruses IV. Because subcutaneous C1498 tumors exhibit growth close to the musculature and aggressively infiltrate into the peritoneal cavity, thus making caliper measurements inaccurate, the C1498 tumors were excised at day 30 after tumor cell implantation in all mice and weighed. As shown in Figure 1B, tumor weights in all VSV-mIFNβ-NIS–treated groups were significantly lower than saline controls, indicating that the virus has antitumor activity at all dose levels tested. Higher doses of virus (107 and 108 TCID50) were significantly better at controlling tumor growth than 106 TCID50.
Combination VSV therapy with anti-PD-L1 Ab was tested. Imaging studies showed that anti-PD-L1 Ab did not compromise VSV infection and spread in the tumors (Figure 1A). The efficacy experiment indicated that the anti-PD-L1 Ab (3 doses given 3 days apart) is active compared with its isotope Ab control (P = .0008), and its activity is comparable to 1 low (106 TCID50) dose of VSV-mIFNβ-NIS (Figure 1B). Combination therapy of VSV-mIFNβ-NIS (at all dose levels) with anti-PD-L1 Ab was significantly superior to Ab alone. However, it is only in the low-dose 106 TCID50 VSV group that combination VSV/Ab therapy is superior to VSV alone (P = .0295).
In vitro analysis of VSV and anti-PD-L1–treated tumors
Anti-VSV staining confirmed robust VSV expression in the tumors of mice treated IV with 108 TCID50 VSV, with and without anti-PD-L1 Ab (Figure 2A). Minimal CD8+ T cells were found in the saline or VSV-treated tumors, but increased in the Ab and VSV/Ab-treated group (Figure 2A). The anti-PD-L1 and VSV/anti-PD-L1 groups are not significantly different (P = .28). Because anti-PD-L1 Ab therapy might inhibit VSV propagation by promoting T-cell–mediated clearance of virally infected cells, an infectious virus recovery assay was performed on tumors from mice that received 108 TCID50 VSV. Virus was recovered from the tumors, and there was no significant difference between the VSV with and without anti-PD-L1 Ab (Figure 2B). Tumors harvested from the 106 TCID50 group showed less extensive VSV staining and lower levels of virus recovery (supplemental Figure 2). Immunohistochemical staining showed that there was high PD-L1 expression on tumors harvested from mice that received saline only (Figure 2C). In contrast, PD-L1 expression was undetectable in tumors from mice that received anti-PD-L1 Ab therapy.

Analysis of explanted C1498 tumors revealed robust VSV infection of tumors. C1498 tumors from mice treated IV with 108 TCID50 VSV-mIFNβ-NIS, with and without anti-PD-L1 Ab, were excised on day 3 after VSV therapy. (A) (i) Representative images of immunohistochemical staining for distribution of VSV infectious foci and CD8 T cells in tumors. (ii) The extent of VSV and CD8 staining in tumors (percentage of coverage) was analyzed on NIH ImageJ software. Data represent mean ± SEM (n = 3 mice) from each group. (B) Viral titers recovered from explanted tumors of mice that received 108 TCID50 VSV. The data are expressed as TCID50 virus per mg of tumor (n = 3, mean ± SEM). (C) (i) Immunohistochemical staining showing PD-L1 expression on C1498 tumors, harvested at 3 days post-Ab. Scale bar represents 100 μm. (ii) Histogram and error bars represent the mean ± SEM (n = 3 mice) from each group. tx, treatment.
Analysis of explanted C1498 tumors revealed robust VSV infection of tumors. C1498 tumors from mice treated IV with 108 TCID50 VSV-mIFNβ-NIS, with and without anti-PD-L1 Ab, were excised on day 3 after VSV therapy. (A) (i) Representative images of immunohistochemical staining for distribution of VSV infectious foci and CD8 T cells in tumors. (ii) The extent of VSV and CD8 staining in tumors (percentage of coverage) was analyzed on NIH ImageJ software. Data represent mean ± SEM (n = 3 mice) from each group. (B) Viral titers recovered from explanted tumors of mice that received 108 TCID50 VSV. The data are expressed as TCID50 virus per mg of tumor (n = 3, mean ± SEM). (C) (i) Immunohistochemical staining showing PD-L1 expression on C1498 tumors, harvested at 3 days post-Ab. Scale bar represents 100 μm. (ii) Histogram and error bars represent the mean ± SEM (n = 3 mice) from each group. tx, treatment.
VSV-mIFNβ-NIS and anti-PD-L1 treatment decreases C1498.GFP leukemia burden in vivo
We next evaluated VSV therapy in a murine model of AML. To facilitate identification of the cells, C1498 cells were transduced with a lentivirus vector encoding the enhanced GFP protein (C1498.GFP). Abundant C1498.GFP cells were found in the blood, liver, spleen, and bone marrow 12 days after systemic administration of cells. C57/B6 mice with advanced AML (day 20 post cell implantation) were given an IV dose of 108 VSV-mIFNβ-NIS. Mice were harvested 48 hours later and single-cell suspensions were made from the major organs. As shown in Figure 3A, VSV-mIFNβ-NIS is active against systemic disease; fewer C1498.GFP-positive cells were found in blood, bone marrow, liver, and spleen 2 days post-VSV treatment.

Antitumor activity of VSV in a murine AML C1498.GFP model. (A) C57BL/6 mice were given C1498.GFP IV and, on day 20, received 1 dose 108 TCID50 VSV-mIFNβ-NIS IV; the mice were harvested 48 hours later. Representative photographs show abundant GFP-positive C1498 cells in control mice, but fewer C1498.GFP cells in VSV-treated mice. (B) Single-cell suspensions from the liver and bone marrow were analyzed by flow cytometry after staining with an anti-PD-L1 Ab and gating GFP-positive cells. Bar and error bars (top right) represent the mean ± SEM (n = 4 mice) from each group, showing significantly lower levels of PD-L1 on tumor cells from mice that received the Ab treatment. (C) Kaplan-Meier survival curves of mice in the various treatment groups (n = 11 or 12 mice in each group). Mice received VSV-mIFNβ-NIS IV on day 0, followed 3 days later with anti-PD-L1 Ab or isotype control Ab (3 total doses). The P values are shown. (D) Kaplan-Meier survival curves of mice that received VSV and anti-PD-L1 Ab, with and without depletion of specific immune cell populations (n = 15 mice in each group). Untreated mice (isotype) or mice depleted of CD4, CD8, or NK cells received VSV-mIFNβ-NIS IV on day 0, followed 3 days later with IP injections of 200 μg of anti-PD-L1 Ab. The P values indicating significance level between groups are shown. BM, bone marrow.
Antitumor activity of VSV in a murine AML C1498.GFP model. (A) C57BL/6 mice were given C1498.GFP IV and, on day 20, received 1 dose 108 TCID50 VSV-mIFNβ-NIS IV; the mice were harvested 48 hours later. Representative photographs show abundant GFP-positive C1498 cells in control mice, but fewer C1498.GFP cells in VSV-treated mice. (B) Single-cell suspensions from the liver and bone marrow were analyzed by flow cytometry after staining with an anti-PD-L1 Ab and gating GFP-positive cells. Bar and error bars (top right) represent the mean ± SEM (n = 4 mice) from each group, showing significantly lower levels of PD-L1 on tumor cells from mice that received the Ab treatment. (C) Kaplan-Meier survival curves of mice in the various treatment groups (n = 11 or 12 mice in each group). Mice received VSV-mIFNβ-NIS IV on day 0, followed 3 days later with anti-PD-L1 Ab or isotype control Ab (3 total doses). The P values are shown. (D) Kaplan-Meier survival curves of mice that received VSV and anti-PD-L1 Ab, with and without depletion of specific immune cell populations (n = 15 mice in each group). Untreated mice (isotype) or mice depleted of CD4, CD8, or NK cells received VSV-mIFNβ-NIS IV on day 0, followed 3 days later with IP injections of 200 μg of anti-PD-L1 Ab. The P values indicating significance level between groups are shown. BM, bone marrow.
C57BL/6 mice with disseminated C1498.GFP AML were given VSV-mIFNβ-NIS (108 TCID50) on day 0, followed by IP administrations of isotype control or anti-PD-L1 Ab (200 μg) on days 3, 6, and 9. Kaplan-Meier survival curves were plotted (Figure 3C). Treatment with anti-PD-L1 Ab (P = .0259), VSV-mIFNβ-NIS (P = .0043), or combination VSV and anti-PD-L1 Ab (P < .0001) significantly extended the survival of mice with AML compared with isotype Ab-treated controls. Combination VSV and anti-PD-L1 Ab therapy was significantly superior to anti-PD-L1 Ab alone (P = .0045) or virus alone (P = .0153). Combination therapy did not result in any adverse clinical events or any significant changes in liver function tests (supplemental Figure 3). The bone marrow and liver were harvested from mice and single-cell suspensions were made. Cells were stained with anti-PD-L1 Ab and flow cytometry analysis of PD-L1 levels on gated GFP-positive tumor cells indicated that VSV treatment did not increase PD-L1 levels on the tumor cells (Figure 3B). Lower detectable levels of PD-L1 were seen on C1498 cells from mice that received anti-PD-L1 Ab. This could also be due to steric hindrance of the bound anti-PD-L1 therapeutic Ab, preventing new Ab from detecting PD-L1 on tumor cells or actual downmodulation of PD-L1 levels by Ab treatment.
Immune cell depletion was performed to determine the impact of specific T-cell populations on combination VSV/anti-PD-L1 Ab therapy (Figure 3D). Mice received anti-CD4, anti-CD8, or anti-NK or isotype Abs 3 days after AML cells were given IV. Depletion of the respective T-cell populations was confirmed by flow cytometry analysis of the peripheral blood (data not shown). Mice received saline (control) or 1 dose of VSV-mIFNβ-NIS (108 TCID50; IV) on day 10 post-C1498.GFP cells and anti-PD-L1 Ab (200 μg/mouse) started day 13 post-cell implantation and once every 3 days. Kaplan-Meier survival curves showed that depletion of CD8 or NK cells resulted in loss of antitumor activity and the survival curves were not significantly different from the saline control. Thus, combination VSV/anti-PD-L1 Ab therapy requires CD8 cells and NK cells. Depletion of CD4 cells did not significantly impact VSV/anti-PD-L1 Ab therapy in this model (P = .084).
Tumor burden and CD4 and CD8 immune cell infiltrates at necropsy
Whole-blood and major organs were harvested when the mice in the survival study were euthanized due to disease progression. Single-cell suspensions were prepared and analyzed by flow cytometry for tumor burden, as indicated by presence of GFP-positive tumor cells (Figure 4A). Mice that received VSV and anti-PD-L1 Ab have the lowest percentage of C1498.GFP tumor cells in the blood, bone marrow, and spleen at death (Figure 4B). Overall, these results show that combination therapy with anti-PD-L1 treatment was most effective at reducing tumor burden in the mice (Figure 4B). Only the combination group showed significantly lower tumor burden in the blood, bone marrow, and spleen, when mice had to be removed from the study due to disease progression.

Combination VSV-mIFNβ-NIS and anti-PD-L1 Ab treatment significantly decreases tumor burden in AML mice. Quantitative analysis of percentage of GFP-positive tumor cells in the single-cell preparations. (A) Representative flow data and (B) scatter plots of the percentage of C1498.GFP cells in the cell suspensions. Mean ± SEM is shown (n = 4-6 samples per group). *P ≤ .05. The unpaired Student t test was used. FSC-A, forward scatter.
Combination VSV-mIFNβ-NIS and anti-PD-L1 Ab treatment significantly decreases tumor burden in AML mice. Quantitative analysis of percentage of GFP-positive tumor cells in the single-cell preparations. (A) Representative flow data and (B) scatter plots of the percentage of C1498.GFP cells in the cell suspensions. Mean ± SEM is shown (n = 4-6 samples per group). *P ≤ .05. The unpaired Student t test was used. FSC-A, forward scatter.
It was previously reported that anti-PD-L1 Ab single-agent therapy resulted in enhanced numbers of infiltrating immune cells in the livers of C1498 tumor mice.23 The presence of CD8+ and CD4+ immune cell infiltrates was evaluated using liver samples of these mice (supplemental Figure 4). The percentage of CD8+ cells in the VSV and anti-PD-L1 Ab group was 14.6%, compared with the control group at 2.7% and the Ab-only group at 3.8%. CD8+ T-cell numbers increased significantly in the VSV/anti-PD-L1 group compared with isotype control (P = .0119) and were different from the anti-PD-L1-only group (P = .0287). CD4+ T cells were also significantly increased in the VSV/anti-PD-L1 Ab group. Taken together, these results indicate a heightened T-cell immune response against leukemia cells when the VSV-mIFNβ-NIS was combined with PD-L1 blockade.
VSV and anti-PD-L1 Ab therapy enhanced numbers of virus and tumor-specific CD8 cells
Mice with systemic AML disease were treated with VSV-mIFNβ-NIS (108 TCID50 IV), anti-PD-L1 Ab (2 doses IP, given 3 and 6 days post-VSV), or combination VSV/anti-PD-L1 Ab therapy 10 days post-cell implantation. Due to aggressive progression of disease in untreated animals, all mice were harvested day 17 post-cell implantation. Splenocytes were stimulated with VSV or GFP immune-dominant peptides and analyzed in IFN-γ ELISPOT, pentamer, and IFN-γ intracellular staining flow cytometry assays. As shown in Figure 5, panel A (pentamer assay) and panel B (IFN-γ staining), VSV-nucleocapsid (N)–specific CD8+ T cells were significantly increased in the VSV group compared with the anti-PD-L1 group. The percentage of VSV-N CD8+ cells was enhanced by addition of anti-PD-L1 Ab to VSV therapy. IFN-γ ELISPOT assay also confirmed that the VSV/anti-PD-L1 Ab combination group had the highest level of VSV-N–specific T cells (Figure 5C).

Characterization of T-cell responses against viral and tumor antigens in VSV-treated C1498.GFP tumor bearing mice. Analysis for VSV (A-C) and GFP tumor antigen (D-F) T cells by pentamer assay, IFN-γ intracellular staining, and ELISPOT IFN-γ assay. C57BL/6 mice were given C1498.GFP IV and, on day 10, received 1 dose 108 TCID50 VSV-mIFNβ-NIS IV, with or without anti-PD-L1 Ab on days 13 and 16. Mice were harvested 7 days post-VSV and splenocytes were used in assays to evaluate the presence of virus or tumor-reactive T cells. Splenocytes were stimulated with VSV NP 52 peptide or EGFP118-126 peptides to detect for VSV or GFP-reactive T cells. (A,D) Pentamer assay, (B,E) IFN-γ intracellular staining, and (C,F) ELISPOT IFN-γ assay. Mean ± SEM (n = 3 mice per group). *P ≤ .05. The unpaired Student t test was used. GP, GFP specific peptide (EGFP118–126).
Characterization of T-cell responses against viral and tumor antigens in VSV-treated C1498.GFP tumor bearing mice. Analysis for VSV (A-C) and GFP tumor antigen (D-F) T cells by pentamer assay, IFN-γ intracellular staining, and ELISPOT IFN-γ assay. C57BL/6 mice were given C1498.GFP IV and, on day 10, received 1 dose 108 TCID50 VSV-mIFNβ-NIS IV, with or without anti-PD-L1 Ab on days 13 and 16. Mice were harvested 7 days post-VSV and splenocytes were used in assays to evaluate the presence of virus or tumor-reactive T cells. Splenocytes were stimulated with VSV NP 52 peptide or EGFP118-126 peptides to detect for VSV or GFP-reactive T cells. (A,D) Pentamer assay, (B,E) IFN-γ intracellular staining, and (C,F) ELISPOT IFN-γ assay. Mean ± SEM (n = 3 mice per group). *P ≤ .05. The unpaired Student t test was used. GP, GFP specific peptide (EGFP118–126).
Most importantly, we wanted to evaluate whether VSV-mIFNβ-NIS, a virus that does not encode a tumor antigen, is able to cross-prime CD8 T cells and induce a tumor antigen-specific immune response. The C1498.GFP cells were genetically modified to express GFP to facilitate analysis of tumor burden and, thus, the GFP protein conveniently serves as a tumor-specific antigen in this model. IFN-γ ELISPOT assay showed induction of GFP-specific T cells by VSV (P = .0051) or by anti-PD-L1 Ab (P = .048) compared with the isotype control group. Addition of anti-PD-L1 Ab to VSV in the combination group increased numbers of GFP-specific T cells compared with VSV (P = .032) or anti-PD-L1 alone (P = .0279). The significant increase in GFP-specific CD8 T cells in the VSV/Ab group was confirmed by the pentamer (Figure 5D) and IFN-γ staining (Figure 5E) flow cytometry assays.
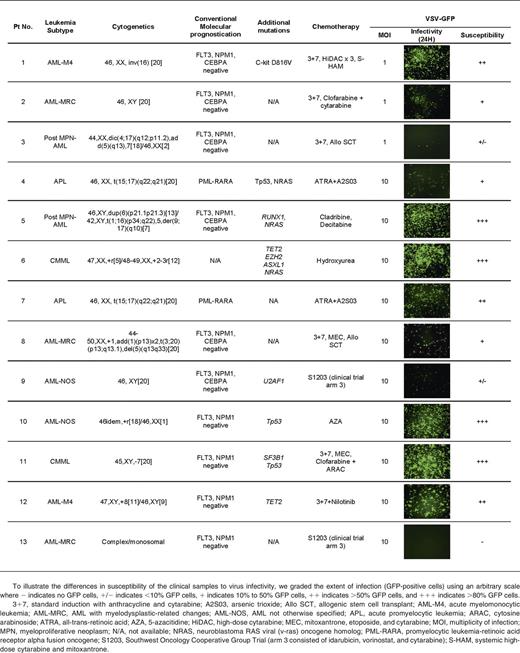
Primary AML cells from clinical samples were susceptible to infection and killing by VSV
Peripheral blood and/or bone marrow MNCs obtained from AML and CMML patients were used immediately after Ficoll separation for virus infection assays. DNA was extracted for genotyping analysis using a 21 gene targeted capture assay. Genes analyzed included FLT3, NPM1, CEBPA, TET2, ASXL1, EZH2, SF3B1, SRSF2, U2AF1, RUNX-1, ETV-6, and CBL. Table 1 demonstrates the clinical, pathological, molecular, and treatment characteristics of the 13 patients. To facilitate rapid screening of cell susceptibility to VSV infection, VSV-GFP was used (Table 1). To illustrate the differences in susceptibility of the clinical samples to virus infectivity, we graded the extent of infection (GFP-positive cells) using an arbitrary scale (Table 1). There was variability in AML/CMML susceptibility to VSV. Overall, ∼50% of the samples received were reasonably susceptible to infection by VSV-GFP. Although the sample size for each specific AML/CMML type is still small, it appears that CMML (2 of 2 cases) and acute myelomonocytic leukemia (AML-M4) (2 of 2 cases) might be especially good targets for VSV therapy.
Discussion
AML is a devastating disease and novel approaches to AML therapy are needed. Here, we show that oncolytic VSV-mIFNβ-NIS has single-agent activity against subcutaneous tumors and disseminated AML in immunocompetent mice. The mechanism of antitumor activity is in part due to VSV infection and oncolytic activity, as revealed by the NIS imaging data, immunohistochemical staining for the VSV antigen showing infectious foci throughout the tumor and virus replication as confirmed by virus recovery assay (about 103 TCID50 per mg tumor). In this model, VSV antitumor activity was enhanced by host adaptive immune responses and requires CD8 and NK cells. VSV oncolysis is followed by immune-mediated antitumor activity with increase in CD4+ and CD8+ T cells filtrating into the tumor and organ in the presence of the checkpoint inhibitor. VSV-mIFNβ-NIS therapy was able to induce cross-priming of CD8 T cells resulting in induction of tumor-specific (GFP) T cells. Importantly, VSV antitumor activity (survival curves and tumor-specific T cells) was boosted in combination with checkpoint blockade using an anti-PD-L1 Ab. Immunohistochemical staining and flow cytometry analysis showed that PD-L1 Ab-treated tumor cells have lower PD-L1 levels. Steric hindrance of the bound anti-PD-L1 therapeutic Ab could have prevented new Ab from detecting PD-L1 on tumor cells. When cultured in vitro, C1498 cells express low/minimal levels of PD-L1 and the levels are upregulated in vivo. Binding of the anti-PD-L1 Ab to PD-L1 could have resulted in downmodulation of PD-L1 from the tumor cell surface. It is also possible that binding of anti-PD-L1 to PD-L1 resulted in Ab-dependent complement-mediated cytotoxic killing of the tumor cells, thereby clearing of high PD-L1–expressing C1498 cells. These 3 possible scenarios remain to be tested in future experiments.
The preclinical study here supports the oncolytic virotherapy paradigm whereby a systemically administered virus extravasates and spreads at tumor sites causing tumor destruction through direct cell killing, followed by immune-mediated clearance of residual tumor cells. The potential of a single-shot therapy for cancer for durable response has been demonstrated in preclinical models, and was recently validated in a phase 1 clinical trial with a single high dose of systemically administrated measles virus encoding the NIS imaging gene.5,8 SPECT/CT reporter gene imaging showed selective infection of disseminated solid plasmacytomas by measles virus encoding NIS, followed by complete remission of these solid plasmacytomas and myeloma disease in the bone marrow.24 In addition to cell killing due to replicative spread of the virus, acute apoptosis in uninfected tumor cells can be induced by antiangiogenesis and antivasculature mechanisms, as shown in oncolytic vaccinia virus therapy.25-27 Oncolytic viruses can also activate innate and adaptive tumor-specific immunity. For some viruses (eg, T-vec, reovirus), this antitumor immune response can sometimes be more important than that of direct oncolysis where direct injection of the primary tumor is associated with regression of distant nodules.28-31 Although cancer cells express tumor antigens, spontaneous rejection of tumors by cytotoxic CD8+ T cells rarely occurs due to the immunosuppressive tumor microenvironment and negative regulatory pathways, including activated cytotoxic T lymphocyte antigen 4 (CTLA-4) and PD-1/PD-L1 immune pathways.32,33 However, cellular responses to the viral infection coupled with release of tumor antigens by virally infected dying tumor cells into the microenvironment attract innate and adaptive immune cells into the tumor, as well as potentially activating resident lymphocytes (including tumor-specific CD4+ and CD8+ T cells).34 This response in association with virus infection makes virotherapy an ideal modality to combine immune checkpoint inhibitors to achieve a more durable and deeper response and outcome.
There is intense academic and industry interest in checkpoint inhibitor Abs that block CTLA-4, PD-1, and PD-L1 for cancer immunotherapy. The most advanced virus/checkpoint inhibitor clinical data are from Amgen where they reported promising early clinical trial data showing improved overall survival of melanoma patients given intratumoral injection of T-vec in combination with ipilimumab (anti-CTLA-4 Ab) compared with T-vec alone.35 CTLA-4 is upregulated on the surface of T cells during the early stages of activation, and downregulates T-cell responses.36 Superior therapeutic outcome was seen with IP delivery of a Her2/neu-targeted oncolytic VSV in combination with an anti-CTLA-4 monoclonal Ab against Her2/neu transgenic murine mammary tumors.37,38 Oncolytic Newcastle Disease virus (NDV) alone has no single-agent activity against B16 tumors in C57/B6 mice, but NDV in combination with CTLA-4 Ab resulted in immune cell infiltration into the tumors and enhanced antitumor activity.39 Combination therapy also led to protection of mice from tumor rechallenges in poorly immunogenic tumors and B16 cells which are refractory to NDV-mediated lysis. In some instances, the oncolytic virus (Adenovirus, measles virus) was engineered to encode anti-CTLA-4 Ab to enhance local concentration of the inhibitor and minimize the immune-related adverse effects associated with systemic anti-CTLA-4 therapy.40-42
In this study, we chose to evaluate the activity of oncolytic VSV-IFNβ-NIS in combination with an anti-PD-L1 inhibitor. There is increasing evidence that the PD-1/PD-L1 pathway (immune checkpoint receptor and ligand) is important in AML. Zhang et al injected the murine AML C1498 cells IV in mice and show that these grew progressively and evaded immune destruction.23 Low levels of PD-L1 expression were found on C1498 cells grown in vitro. However, PD-L1 expression was upregulated on C1498 cells when grown in vivo. VSV-mIFNβ-NIS exhibits potent antitumor activity as a single agent, with a dose-dependent activity (106, 107, 108 TCID50) against subcutaneous C1498 tumors. C1498 is a VSV-permissive model where there is clear evidence of VSV replication with strong VSV antigen staining and infectious virus recovery in VSV-treated tumors. CD8+ infiltrating T cells were seen in the VSV-treated tumors. Combination VSV/anti-PD-L1 Ab treatment did not result in superior activity at the high-dose treatment groups due to permissiveness of the tumors to VSV cell killing. It is in the low-dose treatment group (106 TCID50) that combination therapy was superior to VSV alone. This is highly encouraging as a dose-sparing strategy as it may allow for lower doses of virus to be given to achieve a specific therapeutic effect. Importantly, for viruses that are well tolerated with no adverse effects but not clear therapeutic activity at the highest feasible dose, the data shown here provide a strong rationale to combine the virus with an immune checkpoint inhibitor.
In the systemic AML model, the C1498-GFP cells seed to the bone marrow, liver, and spleen, and eventually in the blood. Systemic VSV therapy resulted in rapid killing of AML cells in these tissues. Forty-eight hours post-VSV, significantly less GFP-positive cells can be found in the spleen, bone marrow, and blood compared with the control. Combination VSV and anti-PD-L1 Ab therapy enhanced survival of mice, and is associated with increase of CD4+CD8+ T cells in the tissues. In this study, VSV was given first, followed by Ab 3 days later for a total of 3 doses. This is to enable maximal tumor cell killing by VSV prior to activation of T-cell responses, which might curtail viral replication by immune cell antiviral responses.
Even though the murine C1498 AML line is highly aggressive but responds well to VSV therapy, this model may not be representative of human leukemias. We also evaluated VSV infectivity on clinical samples from AML/CMML patients who agreed to donate peripheral blood and/or bone marrow for this study. Approximately 50% of the clinical samples demonstrated significant responses to virotherapy. These responses were more pronounced in samples from patients with AML-M4 and CMML. These are myeloid neoplasms with extensive clonal monocytic proliferation. Currently, it is not clear why these neoplasms are especially more permissive to VSV infection. It is likely due to intracellular factors rather than at the cell entry level as the VSV cellular receptor (low-density lipoprotein receptor) is ubiquitously expressed on many cell types.
The immune status of AML patients should be considered in the context of VSV/anti-PD-L1 therapy. Most of the combination chemotherapy regimens used for AML are myelotoxic and cause a temporary lymphodepletion that improves with count recovery. Exceptions include regimens that contain purine nucleoside analogs such as fludarabine and cladribine. Although the lymphodepletion can certainly impact the CD8+ repertoire of the host, secondary to its outcomes with oncolytic virotherapy with or without checkpoint inhibitors, there is some data suggesting a potential benefit from the prior use of cytotoxic agents. These drugs can stimulate both the innate and adaptive arms of the immune system through the following mechanisms: promoting specific rearrangements on dying tumor cells, which render them visible to the immune system; influencing the homeostasis of the hematopoietic compartment through transient lymphodepletion followed by rebound replenishment of immune cell pools; subverting tumor-induced immunosuppressive mechanisms; and exerting direct or indirect stimulatory effects on immune effectors. Some cytotoxic drugs have been shown to induce an immunogenic type of cell death in tumor cells, promoting the maturation of dendritic cells, ultimately resulting in the induction of potent antitumor responses. In future studies, the impact of prior cytotoxic chemotherapy on the immune repertoire of the host and its interaction with responses to viral therapy and checkpoint inhibitors will need to be studied in detail.43
In summary, VSV-mIFNβ-NIS is active against a subcutaneous and disseminated murine model of AML as well as AML samples harvested from peripheral blood or the bone marrow of patients. Combination therapy with anti-PD-L1 Ab boosted VSV activity with no drug-related (VSV or anti-PD-L1) toxicities. This study warrants clinical testing of VSV for AML, either as a single agent or in combination with a checkpoint inhibitor Ab to maximize the chances of a durable response in this devastating and rapidly progressing disease.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Rosa M. Diaz (Mayo Clinic) for advice on the immunologic assays and Marshall Behrens for veterinary help with animal experiments.
This work was supported by National Institutes of Health/National Cancer Institute (NIH/NCI) grant R01 CA175795 and the Margaret Q. Landenberger Research Foundation.
Authorship
Contribution: K.-W.P., S.J.R., and W.S. designed research; W.S. performed research and analyzed data; M.M.P. collected clinical samples and analyzed clinical data; and W.S., M.M.P., A.R., S.J.R., and K.-W.P. drafted the manuscript.
Conflict-of-interest disclosure: K.-W.P. and S.J.R. and Mayo Clinic have a financial interest (equity) in the technology used in this research. The remaining authors declare no competing financial interests.
Correspondence: Kah-Whye Peng, Department of Molecular Medicine, Mayo Clinic, 200 First St SW, Rochester, MN 55905; e-mail: peng.kah@mayo.edu.