

Key Points
High SUMOylation is a clinically relevant vulnerability in AML, associated with higher European LeukemiaNet 2017 risk, poorer survival, and AML-specific mutations.
TAK-981, a new SUMOylation inhibitor, shows nanomolar and immune-independent anti-AML activity, exhibiting synergy with other AML drugs.

Abstract
Acute myeloid leukemia (AML) generally has an unsatisfactory prognosis despite the recent introduction of new regimens, including targeted agents and antibodies. To find a new druggable pathway, we performed integrated bioinformatic pathway screening on large OHSU and MILE AML databases, discovered the SUMOylation pathway, and validated it independently with an external data set (totaling 2959 AML and 642 normal sample data). The clinical relevance of SUMOylation in AML was supported by its core gene expression which is correlated with patient survival, European LeukemiaNet 2017 risk classification, and AML-relevant mutations. TAK-981, a first-in-class SUMOylation inhibitor currently under clinical trials for solid tumors, showed antileukemic effects with apoptosis induction, cell-cycle arrest, and induction of differentiation marker expression in leukemic cells. It exhibited potent nanomolar activity, often stronger than that of cytarabine, which is part of the standard of care. TAK-981’s utility was further demonstrated in in vivo mouse and human leukemia models as well as patient-derived primary AML cells. Our results also indicate direct and cancer cell-inherent anti-AML effects by TAK-981, different from the type 1 interferon and immune-dependent mechanism in a previous solid tumor study. Overall, we provide a proof-of-concept for SUMOylation as a new targetable pathway in AML and propose TAK-981 as a promising direct anti-AML agent. Our data should prompt studies on optimal combination strategies and transitions to clinical trials in AML.
Introduction
Acute myeloid leukemia (AML) is a heterogeneous disease characterized by an accumulation of immature progenitor cells with arrested differentiation leading to suppression of hematopoiesis.1 Current standard of care treatments include combination chemotherapy with cytotoxic drugs, usage of hypomethylating agents, and/or hematopoietic stem cell transplantation.2 Recent improvement in our understanding of AML pathogenesis has led to the introduction of several novel targeted agents since 2017.3 Nevertheless, long-term survival is still suboptimal without allogeneic hematopoietic stem cell transplantation,4 and thus, more efforts should be done to unravel novel prognostic, predictive, and targetable molecular abnormalities. However, lack of prevailing driver genomic mutations and available unique markers for AML has made it quite difficult. In this context, investigations into postgenomic pathways relevant to AML pathogenesis and approaches to their targeting have been desired.
SUMOylation is a posttranslational modification involved in the conjugation of small ubiquitin-like modifiers (SUMOs) to substrate proteins.5 SUMO-activating enzyme E1 (SAE1 and SAE2 encoded by SAE1 and UBA2, respectively), an E2 (ubiquitin-conjugating enzyme 9 (UBC9) encoded by UBE2I), and a limited set of E3 ligases participate in this process.5,6 SUMOylation seems to be important in the nuclear functions of proliferating or developing cells by regulating the mitotic cell cycle and DNA damage response.7-9 Specific pathways affected by SUMOylation in cancer may include p5310,11 and cMYC,12,13 but more studies are needed to resolve some of the controversies.14,15 In addition, innate immunity is mostly suppressed by SUMOylation, the inhibition of which, therefore, might have implications for cancer therapy.5,16 As for AML, only a few studies on the roles of SUMOylation have been published.17-19 Therefore, concrete evidence of the therapeutic utility of SUMOylation or of specific inhibitors of SUMOylation in AML has been lacking. TAK-981 is an inhibitor of the SUMO-activating enzyme (SAE) that forms a SUMO-TAK-981 adduct.20 As the first-in-class SAE inhibitor targeting cancers, it is currently in clinical trials for solid tumors or lymphomas (#NCT03648372, #NCT04074330, and #NCT04381650). In blood cancer, it has been known to shift the T-cell balance toward healthy immune cell subsets in chronic lymphocytic leukemia.21 To our knowledge, TAK-981 has not been studied for AML or evaluated in AML clinical trials.
For solid tumors, large-scale bioinformatic analysis has been successfully performed comparing normal and cancer samples, thanks to The Cancer Genome Atlas (TCGA) data. TCGA also contains data on AML (TCGA-LAML22 data set), but it lacks the data for noncancer controls, limiting its application in the AML field. As of now, 3 large-scale gene expression databases contain both AML and normal data: (1) MILE study stage I data,23 (2) OHSU data from the Beat AML 1.0 program,24 and (3) the Gene Expression Omnibus (GEO) compilation.25 Therefore, analysis of these large databases (totaling 2959 AML and 642 normal samples) might yield new and useful information on targets for patients with broader AML.
Here, accessing large gene expression databases for AML, we evaluated the clinical relevance of the SUMOylation pathway and investigated the antileukemic effects of its inhibition by TAK-981.
Methods
More detailed information is in supplemental Methods.
Bioinformatic analysis
Details are provided in supplemental Data.
Cells - reagents, antibodies for flow cytometry, and cell viability with cell counting kit-8 (CCK-8) assay
Details are provided in supplemental Data.
Primary AML cells from patients
The information of the samples, including mutation status, is listed in supplemental Table 7.
Flow cytometry, Apoptosis analysis, and Cell-cycle analysis
Details are provided in supplemental Data.
Quantitative reverse transcription polymerase chain reaction validation, Western blotting
The efficiencies of the primers used are listed in supplemental Table 8.
Animal experiments
Details are provided in supplemental Data.
Statistical analysis
The Wilcoxon rank-sum test, one-way analysis of variance, Student t test, and Jonckheere-Terpstra test were used when necessary. Details are provided in supplemental Data.
All animal experiments were performed in accordance with a protocol approved by the Institutional Animal Care and Use Committee of The Catholic University of Korea (CUMC-2020-0318-01). Bone marrow (BM) samples from patients with AML were collected during routine diagnostic procedures after informed consent was obtained in accordance with Institutional Review Board regulations of The Catholic University of Korea (KC20SISI0957) and the Declaration of Helsinki.
Results
Bioinformatic screening identifies SUMOylation pathway as AML-specific target
First, we performed an integrated analysis on large-scale databases (MILE study stage I and OHSU Beat AML 1.0 program) (Figure 1A). Selection of significant pathways in the 2 gene set enrichment analysis results (AML vs normal) (supplemental Table 1) followed by their clustering based on common leading-edge genes and protein-protein interactions yielded 4 distinct pathway clusters: (1) translation/ribosomal RNA/mitochondria, (2) histone-related, (3) SUMOylation, and (4) regulation of messenger RNA (mRNA) (Figure 1B). Interestingly, inhibitors targeting the first cluster, such as ribosome biogenesis inhibitors or tetracyclines, had shown both in vitro and in vivo antileukemic activities and were entered into clinical development.27-29 These facts show that our bioinformatic results may have real relevance for targeting AML. Of the 3 remaining clusters, we focused on SUMOylation cluster because it had not been explored much for AML, and the other 2 were either difficult to establish the causality (Histone-related) or too nonspecific (Regulation of mRNA). Most of the individual genes comprising the SUMOylation pathway were found to be upregulated in AML samples from both the MILE and OHSU databases (SUMO1 and UBA2 in Figure 1C; all the others in supplemental Figure 1A). We further validated the results using another large independent data set from the GEO collection of 2213 AML and 548 normal samples.25 Consistently, we found that 11 of 17 genes related to SUMOylation were found to be significantly upregulated in AML samples (SUMO1 and UBA2 in Figure 1D; all the others in supplemental Figure 1B). In particular, we observed higher protein levels of E1 (SAE1 and SAE2), targets for TAK-981, and E2 (UBC9) in cells from patient with AML than those in healthy control or patients with remission after therapy (Figure 1E). We believe these provide further support for the involvement of SUMOylation at the protein level. The results also suggest that the upregulated SUMOylation pathway in AML may be a target for therapeutic intervention.

Bioinformatic screening to find AML-specific pathways. (A) Overall strategy for database screening. (B) Graphical illustration of 4 pathway clusters upregulated in AML BM samples from panel A, using GSCluster26 R package. The number of connected gene sets in each cluster is indicated. (C) Comparison of UBA2 and SUMO1 gene expression between healthy and AML BM samples in OHSU and MILE databases. (D) Comparison of UBA2 and SUMO1 gene expression between healthy and AML BM/peripheral blood samples in Gene Expression Omnibus (GEO) data sets by Roushangar and Mias.25 (E) Representative western blot for SAE2, SAE1, UBC9, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) in peripheral blood from healthy controls and patients with AML at diagnosis or remission state after treatment (left). The intensities of the bands from all the samples were quantified by densitometry and displayed as the ratio of each protein to GAPDH (loading control) (right). Newly diagnosed patients with AML (n = 7), those at remission state (n = 5), and healthy controls (n = 5). Results are expressed as the mean ± standard error of the mean. For panels C-E, P values are from Wilcoxon rank-sum test; ∗P < .05, ∗∗P < .01, ∗∗∗P < .001, ∗∗∗∗P < .0001. BM, bone marrow; GEO, Gene Expression Omnibus; FDR, false discovery rate; GSEA, gene set enrichment analysis; rRNA, ribosomal RNA.
Bioinformatic screening to find AML-specific pathways. (A) Overall strategy for database screening. (B) Graphical illustration of 4 pathway clusters upregulated in AML BM samples from panel A, using GSCluster26 R package. The number of connected gene sets in each cluster is indicated. (C) Comparison of UBA2 and SUMO1 gene expression between healthy and AML BM samples in OHSU and MILE databases. (D) Comparison of UBA2 and SUMO1 gene expression between healthy and AML BM/peripheral blood samples in Gene Expression Omnibus (GEO) data sets by Roushangar and Mias.25 (E) Representative western blot for SAE2, SAE1, UBC9, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) in peripheral blood from healthy controls and patients with AML at diagnosis or remission state after treatment (left). The intensities of the bands from all the samples were quantified by densitometry and displayed as the ratio of each protein to GAPDH (loading control) (right). Newly diagnosed patients with AML (n = 7), those at remission state (n = 5), and healthy controls (n = 5). Results are expressed as the mean ± standard error of the mean. For panels C-E, P values are from Wilcoxon rank-sum test; ∗P < .05, ∗∗P < .01, ∗∗∗P < .001, ∗∗∗∗P < .0001. BM, bone marrow; GEO, Gene Expression Omnibus; FDR, false discovery rate; GSEA, gene set enrichment analysis; rRNA, ribosomal RNA.
SUMOylation pathway is associated with adverse risk features and poor survival in AML
We then explored the clinical relevance of SUMOylation. First, higher expression of most of the important genes in the SUMOylation pathway from the OHSU database (SUMO1 and UBA2 in Figure 2A; all the others in supplemental Figure 2A) was significantly associated with shorter survival. Some of those negative correlations (for SAE1, BMI1, and PHC2) were validated with the TCGA database (supplemental Figure 2A), and all the results, along with those without correlations, are shown in supplemental Table 2. Second, the ELN2017 risk analysis on the 4 groups (healthy, favorable, intermediate, and adverse in OHSU database) demonstrated that most of the core genes in the SUMOylation pathway expressed at higher levels in the high-risk groups (P < .05) (SUMO1, UBA2, and SAE1 in Figure 2B; all the others in supplemental Figure 2B). Post hoc analysis showed that the difference concerning the SUMOylation pathway between the healthy and adverse risk groups was significant (except for UBE2I gene). This trend was also confirmed from the 3 patient risk groups (favorable, intermediate, and adverse) in the TCGA database for several genes, including BMI1, CBX2, and core genes such as SAE1 and UBA2, and the results are shown in supplemental Table 3 along with the results for all the other genes without such confirmation.30 As the above results are for individual gene levels, we further explored the pathway-specific relationship between SUMOylation and overall survival/ELN2017, by performing similar analyses with gene set variation analysis (GSVA) pathway scores.31 Consistent with the results from individual genes, higher scores of SUMOylation pathways were found to be significantly related with a poorer prognosis in both the survival analysis and the ELN2017 risk analysis (supplemental Table 4). These relationships remained valid after adjusting for characteristics of patients with high-risk AML that might have confounding effects, as evidenced by multivariate analysis (supplemental Table 5).

Clinical relevance of SUMOylation pathway in AML. (A) Kaplan-Meier curves with 95% confidence intervals (dotted lines) for overall survival of patients with AML in OHSU, according to the gene expression levels of UBA2 or SUMO1. The division of the high- and low-expression groups was determined by the best risk separation approach. (B) Comparison of UBA2, SUMO1, and SAE1 gene expression across healthy and ELN2017 risk groups. P values are from Jonckheere-Terpstra test. Subsequent post hoc analyses were performed with the two-stage linear step-up procedure, and the significance is indicated for each comparison. The number of participants is indicated for each group. (C) Comparison of UBE2I and SUMO1 gene expression between mutated and wild-type of NPM1, TP53, and RUNX1 genes in OHSU database. P values are from Wilcoxon rank-sum test. For panels B-C, ∗P < .05, ∗∗P < .01, ∗∗∗P < .001, ∗∗∗∗P < .0001. Adv, adverse; Fav, favorable; HR(high), hazard ratio of high expression group; Int, intermediate; ns, not significant.
Clinical relevance of SUMOylation pathway in AML. (A) Kaplan-Meier curves with 95% confidence intervals (dotted lines) for overall survival of patients with AML in OHSU, according to the gene expression levels of UBA2 or SUMO1. The division of the high- and low-expression groups was determined by the best risk separation approach. (B) Comparison of UBA2, SUMO1, and SAE1 gene expression across healthy and ELN2017 risk groups. P values are from Jonckheere-Terpstra test. Subsequent post hoc analyses were performed with the two-stage linear step-up procedure, and the significance is indicated for each comparison. The number of participants is indicated for each group. (C) Comparison of UBE2I and SUMO1 gene expression between mutated and wild-type of NPM1, TP53, and RUNX1 genes in OHSU database. P values are from Wilcoxon rank-sum test. For panels B-C, ∗P < .05, ∗∗P < .01, ∗∗∗P < .001, ∗∗∗∗P < .0001. Adv, adverse; Fav, favorable; HR(high), hazard ratio of high expression group; Int, intermediate; ns, not significant.
Third, we tested if particular gene mutations are related to core SUMOylation gene expression. Among the 4 gene mutations (FLT3-ITD, NPM1, TP53, and RUNX1) that had enough patients (n > 5) for both mutated and wild-type groups, 3 mutations (NPM1, TP53, and RUNX1) exhibited consistent patterns between prognosis and core SUMOylation gene expression (SUMO1 and UBE2I in Figure 2C; all the others in supplemental Figure 2C). Specifically, patients with the NPM1 mutation associated with better prognosis had lower SUMOylation gene expression, whereas those with the TP53 and RUNX1 mutations associated with poor prognosis had higher SUMOylation gene expression. These results suggest that activation of the SUMOylation pathway is associated with adverse risk features and poorer survival.
TAK-981, a new SUMOylation inhibitor, exhibits potent antileukemic effects in vitro
In our quest for an inhibitor of SUMOylation, we found TAK-981, which was developed very recently as a first-in-class inhibitor of SAE step20 and is currently under clinical trial for various solid tumors. As its effects against AML are still unknown, we evaluated them in vitro.
Surprisingly, TAK-981 showed greater or similar potency compared with cytarabine (Ara-C), a standard drug used in clinics, against 4 AML cell lines (Figure 3A). Notably, the 50% inhibitory concentration (IC50) values for TAK-981, all within a 2-digit nanomolar range, were somewhat uniform across the cell lines. By contrast, those for cytarabine differed markedly (>1 μM for KG-1 and THP-1; 2-digit nM range for U937). In comparison, tetracycline, targeting the “translation/ribosomal RNA/mitochondria” identified above, exhibited only several-hundred-μM potency (supplemental Figure 3A).

TAK-981's potency and its synergy with cytarabine for AML cells. (A) Dose-response curves of TAK-981 and cytarabine for 4 AML cell lines. The concentration values right beside each curve represent IC50 values. (B) Synergy between TAK-981 and cytarabine for 4 AML cell lines. (C) Combination index plots computed from the data in panel B by CompuSyn software. (D) Synergy between TAK-981 and several drugs for MOLM-14 cell line. (E) Combination index plots computed from the data in panel D by CompuSyn software. For panels B and D, different concentration ranges were used for each drug, and the error bars indicate standard deviation. For panels C and E, values below the dotted line at 1.0 indicate synergy. For panels A, B, and D, cell viability was measured by CCK-8 assay. Aza, azacitidine; Fa, fractions affected; Qui, quizartinib; TAK, TAK-981; Ven, venetoclax.
TAK-981's potency and its synergy with cytarabine for AML cells. (A) Dose-response curves of TAK-981 and cytarabine for 4 AML cell lines. The concentration values right beside each curve represent IC50 values. (B) Synergy between TAK-981 and cytarabine for 4 AML cell lines. (C) Combination index plots computed from the data in panel B by CompuSyn software. (D) Synergy between TAK-981 and several drugs for MOLM-14 cell line. (E) Combination index plots computed from the data in panel D by CompuSyn software. For panels B and D, different concentration ranges were used for each drug, and the error bars indicate standard deviation. For panels C and E, values below the dotted line at 1.0 indicate synergy. For panels A, B, and D, cell viability was measured by CCK-8 assay. Aza, azacitidine; Fa, fractions affected; Qui, quizartinib; TAK, TAK-981; Ven, venetoclax.
Next, we tested TAK-981 for any synergistic or dose reduction effect when used with cytarabine in the 4 cell lines (Figure 3B-C). In addition, TAK-981’s synergy with 2 new targeted-therapy drugs, venetoclax and quizartinib, along with a demethylating drug, azacitidine, was tested for the MOLM-14 cell line having the FLT3-ITD mutation, which is associated with poor prognosis (Figure 3D-E). Synergy, as judged by the CompuSyn scores,32 varied substantially across cell lines, with U937 and MOLM-14 exhibiting significant synergy, whereas KG-1 and THP-1 showing little synergy in the combination with cytarabine. For MOLM-14, TAK-981 exhibited significant synergy with azacitidine, some synergy at higher drug concentrations with venetoclax, but no synergy with quizartinib. In addition, TAK-981 showed similar and lower potency in comparison with venetoclax, a BCL2 inhibitor, and quizartinib, an FLT3 inhibitor, respectively (Figure 3D; supplemental Figure 3B). Although we used only concentration values around IC50 for each drug, significant synergy might be observed with different concentration combinations. We also assessed the dosage reduction effects of TAK-981 (Table 1). Notably, even when there was no apparent synergy, the dose reduction indices of the drugs combined with TAK-981 were above 1 for all of the drug cell line settings, indicating significant dosage reduction effects. This could be exploited to lower the toxicity of such drugs when combined with TAK-981. Overall, TAK-981’s combination with conventional or targeted drugs holds promise for improved therapeutics.
Dose reduction index of cytarabine or other drugs when combined with TAK-981 in AML cell lines
| Cell line | Combination drug | Dose reduction index at fraction affected (Fa) = 0.9 |
|---|---|---|
| U937 | Cytarabine | 12.03 |
| THP-1 | Cytarabine | 1.25 |
| KG-1 | Cytarabine | 2.61 |
| MOLM-14 | Cytarabine | 2.94 |
| MOLM-14 | Azacitidine | 4.87 |
| MOLM-14 | Quizartinib | 8.64 |
| MOLM-14 | Venetoclax | 4.98 |
| Cell line | Combination drug | Dose reduction index at fraction affected (Fa) = 0.9 |
|---|---|---|
| U937 | Cytarabine | 12.03 |
| THP-1 | Cytarabine | 1.25 |
| KG-1 | Cytarabine | 2.61 |
| MOLM-14 | Cytarabine | 2.94 |
| MOLM-14 | Azacitidine | 4.87 |
| MOLM-14 | Quizartinib | 8.64 |
| MOLM-14 | Venetoclax | 4.98 |
Fa = 0.9 refers to the point where the inhibition effect is 90%, that is, when 90% of the cells are dead. The number 0.9 was chosen, because for cancer therapies, high effect levels are thought to be more therapeutically relevant than low effect levels.32
TAK-981 induces apoptosis, cell-cycle arrest, and/or differentiation marker expression in AML cell lines
To study how TAK-981 exhibits antileukemic effects, we investigated cellular events upon drug treatment. As expected, TAK-981 reduced SUMOylation for some of the proteins, if not all, from the cell extracts (24- or 48-hour treatment) (Figure 4A; supplemental Figure 4). Because SUMOylation plays a critical role in transcription regulation, we next analyzed gene expression profile changes by TAK-981 treatment (16 hours) using gene set enrichment analysis (GSE173116:33 THP-1 cells) (Figure 4B). The upregulated pathways included those for cell death and cell-cycle arrest, such as the p53 pathway and apoptosis. Experimentally, the mRNA expression of genes for apoptosis (DDIT3) and cell-cycle arrest (P21 and TP53), known to be downregulated by SUMOylation in AML cells,11,19,34 were significantly higher in TAK-981-treated THP-1 cells (48 hours) than in those from the control or cytarabine–treated group (Figure 4C). We also found that there was a trend that SUMO core pathway is downregulated in TAK-981–treated THP-1 cells (supplemental Figure 5), although TAK-981’s posttranslational effect on SUMO may not necessarily involve the expression of SUMO core genes. Further analysis in several other AML cell lines with western blot (p21, caspase 3, and cytochrome C) (Figure 4D), flow cytometry for apoptosis (Figure 4E), and DNA content analysis (Figure 4F) showed that apoptosis and cell-cycle arrest were generally observed for the TAK-981–treated AML cells (48 hours), with only minor variations. For example, G2/M phase arrest was observed for U937, THP-1, and KG-1 cells, whereas G0/G1 arrest was observed in MOLM-14 cells. Meanwhile, there is heterogeneity in terms of p53 mutations among the cell lines used in this study (supplemental Table 6). As p21 can be regulated either by p53 dependently or independently, we tested if the induction of p21 by TAK-981 is also reflected in the p53. TAK-981 treatment did not change the levels of either p53 or MDM2 (supplemental Figure 6), suggesting that the TAK-981–induced p21 change may not be related to p53. Possible mechanistic disconnection between p53 and p21 upon TAK-981 treatment could be an interesting topic for future research.
Apoptosis, cell-cycle arrest, and differentiation induced by TAK-981 in AML cells. (A) Effect of TAK-981 on protein SUMOylation in AML cells after 24- (U937, THP-1) or 48-hour (MOLM-14, KG-1) treatment. Western blot analysis was performed with the antibody for SUMO-2/3/4. (B) Top 12 pathways with P < .05 from GSEA analysis of TAK-981–treated THP-1 cells from GSE173116 data set with the Hallmark gene set (left). The pathways are in the order of the normalization of the enrichment score (NES). Enrichment score plots for genes belonging to p53 and apoptosis pathways from the GSEA analysis (right). (C) Relative mRNA expression of DDIT3, P21, and TP53 in TAK-981 (indicated concentrations) and cytarabine (1 μM) in THP-1 cells after 48-hour treatment, as measured by quantitative reverse transcription polymerase chain reaction. (D) Western blot for p21, cleaved caspase-3, and cytochrome C expression in AML cells after 48-hour treatment with TAK-981. (E) Apoptosis analysis for TAK-981–treated AML cells (48 hours) by flow cytometry with Annexin V/propidium iodide (PI) kit. Apoptotic cells (%) (right) is the sum of the early (Q3) and late (Q2) apoptosis percentages. (F) Cell-cycle analysis for TAK-981–treated AML cells after 48 hours by flow cytometry. Each phase of cell cycle was analyzed with cell-cycle platform in FlowJo software. Quantitative reverse transcription polymerase chain reaction analysis of differentiation markers and CD39 gene. mRNA expression in 48-hour TAK-981–treated AML cells for CD15 in U937, CD14 in THP-1, and CD11B in MOLM-14 (G) and CD39 in all cells (H). Two-tailed Student t test was used for panels C, E, G, and H, and one-way analysis of variance was used for panel F. Data are expressed as mean ± standard deviation (n = 3); ∗P < .05, ∗∗P < .01, ∗∗∗P < .001, ∗∗∗∗P < .0001. For all except panel B, the drug concentrations were selected so that TAK-981 did not kill all the cells but had detectable effects on cells, based on the results from the initial estimation of IC50 of TAK-981 for each cell line. The time points are different because the emergence of the particular effects was expressed at different time points according to cell lines. In addition, all experiments were done with n = 3. For panels C, G, and H, the expression values were normalized against that of β-actin (ACTB). The efficiencies of the primers used are listed in supplemental Table 8. Ara-C, cytarabine; IL6, interleukin 6; JAK, Janus kinase; NF-κB, nuclear factor kappa B; STAT, signal transducer and activator of transcription; TGF, transforming growth factor; TNF, tumor necrosis factor.

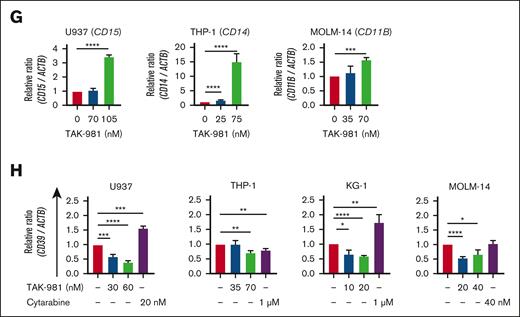
TAK-981 treatment (48 hours) also affected the differentiation of leukemic cells dose-dependently, as shown by the increase in the differentiation markers for U937 (CD15),34-36 THP-1 (CD14), and MOLM-14 (CD11B) cells (Figure 4G). Moreover, TAK-981 suppressed the expression of CD39 (48 hours) (Figure 4H), which is known to be involved in AML chemoresistance,37 in both chemosensitive (U937) and chemoresistant cells (KG-1, THP-1, and MOLM-14). These data suggest that TAK-981 exhibits antileukemic effects by inducing apoptosis, cell-cycle arrest, differentiation, or lower chemoresistance.
TAK-981 potency in primary AML cells ex vivo
The effects of TAK-981 were also evaluated ex vivo in primary AML cells from patient B (n = 13). TAK-981 exhibited higher inhibition of primary cell proliferation at equimolar concentrations than did cytarabine, which did not appreciably inhibit the cells at up to ∼50 μM concentrations (Figure 5A). Interestingly, the inhibitory potencies of both compounds for the primary cells were much lower than those for the AML cell lines. In addition, the SUMOylation status of primary AML cells from patients was lower than that in the cell lines (supplemental Figure 7). The possible reasons for these differences between cell lines and primary cells are addressed in the discussion section.
![TAK-981’s activity against primary AML cells ex vivo. Freshly isolated mononuclear cells from BM of 13 patients with AML were cultured with different doses of TAK-981, cytarabine, or both for 48 hours. Leukemic cells were gated with CD33 and/or CD34 by flow cytometry and viable cells (4′,6-diamidino-2-phenylindole [DAPI]–negative/Annexin V–negative) were compared between groups. (A) Potency and combination effects of TAK-981 and cytarabine. Viable cells were estimated by flow cytometric analysis of primary AML cells treated with TAK-981, cytarabine, or both. Error bars are standard errors. (B) Synergistic combination index between TAK-981 and cytarabine from data in panel A. (C) Leukemic cell gating (left) and representative data of flow cytometry for apoptosis of primary AML cells at different concentrations of TAK-981 with DAPI and Annexin V.](/view-large/figure/11758973/BLOODA_ADV-2022-007956-gr5.jpg)
TAK-981’s activity against primary AML cells ex vivo. Freshly isolated mononuclear cells from BM of 13 patients with AML were cultured with different doses of TAK-981, cytarabine, or both for 48 hours. Leukemic cells were gated with CD33 and/or CD34 by flow cytometry and viable cells (4′,6-diamidino-2-phenylindole [DAPI]–negative/Annexin V–negative) were compared between groups. (A) Potency and combination effects of TAK-981 and cytarabine. Viable cells were estimated by flow cytometric analysis of primary AML cells treated with TAK-981, cytarabine, or both. Error bars are standard errors. (B) Synergistic combination index between TAK-981 and cytarabine from data in panel A. (C) Leukemic cell gating (left) and representative data of flow cytometry for apoptosis of primary AML cells at different concentrations of TAK-981 with DAPI and Annexin V.
TAK-981’s activity against primary AML cells ex vivo. Freshly isolated mononuclear cells from BM of 13 patients with AML were cultured with different doses of TAK-981, cytarabine, or both for 48 hours. Leukemic cells were gated with CD33 and/or CD34 by flow cytometry and viable cells (4′,6-diamidino-2-phenylindole [DAPI]–negative/Annexin V–negative) were compared between groups. (A) Potency and combination effects of TAK-981 and cytarabine. Viable cells were estimated by flow cytometric analysis of primary AML cells treated with TAK-981, cytarabine, or both. Error bars are standard errors. (B) Synergistic combination index between TAK-981 and cytarabine from data in panel A. (C) Leukemic cell gating (left) and representative data of flow cytometry for apoptosis of primary AML cells at different concentrations of TAK-981 with DAPI and Annexin V.
Still, there was significant synergy between the 2 drugs against the primary cells (Figure 5B), indicating the possible clinical utility of TAK-981. Consistently with the AML cell line results, TAK-981 induced apoptosis in the primary AML cells, and this result suggests its direct effect on cancer cells independent of antitumor immunity (Figure 5C).
TAK-981's antileukemic effects in both syngeneic AML mouse and human xenograft models
To assess TAK-981’s anti-AML activity in an immune-competent environment, we used the mouse syngeneic AML model using the C1498 cell line. For the mice injected with C1498/luciferase/CD90.1 cells through tail veins, TAK-981 significantly reduced the leukemic burden on day 19 relative to the control group, as judged by the bioluminescence (supplemental Figure 8A-B). Flow cytometric analysis of leukemic cells from BM and blood (from 3 euthanized animals from each group on day 19) showed much fewer leukemic cells in the TAK-981 group (supplemental Figure 8C-D), consistent with the above imaging data on day 19. Significantly prolonged survival was also observed in the TAK-981 group relative to the controls (supplemental Figure 8E). These data in the syngeneic, immune-competent cancer model confirm TAK-981’s in vivo anti-AML activity.
To confirm the human relevance of the antileukemic activity of TAK-981 and to evaluate the influence of antitumor immunity on its anti-AML effect, we injected human AML cell MOLM-14/luciferase/green fluorescent protein (0.5 × 106) into nonirradiated, immune-deficient NOD/SCID/IL-2rγnull (NSG) mice (no T cells and defective dendritic cells). Both the bioimaging data (Figure 6A-B) and the flow cytometric results on the blood and BM cells (Figure 6C-D) confirmed the lower leukemic burden in the TAK-981 group. Western blot with sorted leukemic cells showed a decreased level of SUMOylated proteins in the TAK-981 group, thereby confirming its in vivo deSUMOylation activity (Figure 6E). Significantly prolonged survival was also observed in the TAK-981 group relative to the control (Figure 6F). Therefore, the data confirm TAK-981’s anti-human AML activity in vivo. Importantly, these data show that TAK-981’s in vivo activity is independent of antitumor immunity, as it is lacking in the NSG mouse model.

TAK-981's antileukemic effects in human xenograft AML mouse models (immune-compromised mice). (A-E) Human AML mouse model was established by injecting MOLM-14 cells labeled with luciferase/green fluorescent protein (GFP) (MOLM-14/luciferase/GFP) into NSG mice through tail vein. After confirming leukemia engraftment by bioluminescence imaging, the mice were divided into 2 groups (10 mice per group) and treatment began on day 5 until day 26: control (no treatment) or TAK-981 (7.5 mg/kg formulated in 20% 2-hydroxypropyl-β-cyclodextrin, IV 3 times a week). Representative mice from each group were subjected to serial bioluminescence images (A) and intensity quantitation on days 5, 12, and 20 after leukemic cell injection (B). (C-D) Three representative mice per group were euthanized on day 20 to compare the leukemic burdens between the groups. Cells from the BM and blood were analyzed using flow cytometry. The proportions of GFP+ cells by flow cytometry to identify leukemic cells were compared between the groups. (E) Western blot was performed with sorted leukemic cells to evaluate SUMOylated proteins in each group. The sample was pooled from individual animals, representing the average levels (supplemental Methods). (F) The overall survival rate in each group (7 mice per group) was estimated by the Kaplan-Meier method. The results are expressed as the mean ± standard error of the mean; ∗P < .05, ∗∗P < .01. CTL, control; SSC, side scatter.
TAK-981's antileukemic effects in human xenograft AML mouse models (immune-compromised mice). (A-E) Human AML mouse model was established by injecting MOLM-14 cells labeled with luciferase/green fluorescent protein (GFP) (MOLM-14/luciferase/GFP) into NSG mice through tail vein. After confirming leukemia engraftment by bioluminescence imaging, the mice were divided into 2 groups (10 mice per group) and treatment began on day 5 until day 26: control (no treatment) or TAK-981 (7.5 mg/kg formulated in 20% 2-hydroxypropyl-β-cyclodextrin, IV 3 times a week). Representative mice from each group were subjected to serial bioluminescence images (A) and intensity quantitation on days 5, 12, and 20 after leukemic cell injection (B). (C-D) Three representative mice per group were euthanized on day 20 to compare the leukemic burdens between the groups. Cells from the BM and blood were analyzed using flow cytometry. The proportions of GFP+ cells by flow cytometry to identify leukemic cells were compared between the groups. (E) Western blot was performed with sorted leukemic cells to evaluate SUMOylated proteins in each group. The sample was pooled from individual animals, representing the average levels (supplemental Methods). (F) The overall survival rate in each group (7 mice per group) was estimated by the Kaplan-Meier method. The results are expressed as the mean ± standard error of the mean; ∗P < .05, ∗∗P < .01. CTL, control; SSC, side scatter.
Discussion
SUMOylation has not been much recognized in AML other than in cases of acute promyelocytic leukemia (APL), a minor (∼10%) subset of AML with the characteristic chromosomal translocation generating the PML-RARα fusion protein.38 The established therapy for APL, with all-trans retinoic acid (ATRA) and As2O3, triggers SUMOylation and subsequent proteasomal degradation of PML-RARα, thus inducing APL differentiation.39 Activities of ATRA-induced differentiation on some non-APL AML cell lines in vitro40 led to clinical trials, but yielded overall disappointing outcomes.41 In our results, TAK-981 could enhance the in vitro differentiation of all AML cells tested. It will be interesting to revisit the issue of the differentiation of AML cells upon inhibition of SUMOylation in vivo. It is therefore worth noting that the addition of ATRA to decitabine improved clinical outcomes for older patients who are difficult to treat in a phase 2 clinical trial.42 There have also been a few reports on the SUMOylation of individual proteins involved in AML, such as iGF1R, sPRDM, and ERG.17,18,43 In addition, a protein array–based screening on AML cell lines with acquired drug resistance vs parental cell lines identified possible SUMOylation biomarkers related to drug resistance, which is yet to be validated in vivo.44 However, considering the inhibition of the initial step of SUMOylation by TAK-981, it seems unlikely that one particular protein is responsible for TAK-981’s antileukemic activity. Rather, TAK-981’s activity should be contributed to by several SUMOylation-dependent processes.45 The differential profiles of SUMOylation dependency might explain why we observed a large variability in synergy between TAK-981 and cytarabine across the different AML cells. Inhibition of SUMOylation in general with different inhibitors has also been tested. Anacardic acid and/or 2-D08 induce apoptosis of leukemic cells through reactive oxygen species–mediated deSUMOylation of NOX or DDIT3 regulators.19,46 In addition, anacardic acid and 2-D08 sensitized non-APL AML cells to ATRA-based differentiation.34 However, there is a conflicting report according to which anacardic acid and ginkgolic acid alleviated ATRA-mediated inhibition of leukemic cell proliferation.47 This shows that SUMOylation inhibition for AML therapy has not yet been well established and that the existing literature may need to be considered with some caution. Particularly, most of these studies have used cell lines in vitro or subcutaneous flank xenografts of AML cells and inhibitors with rather moderate micromolar activities without high specificity for SUMOylation.34,47 In comparison, we started from the clinical relevance of the SUMOylation pathway and investigated the association of core genes in the SUMOylation pathways and AML characteristics, rather than focusing on a single protein. Furthermore, we evaluated a highly specific SUMOylation inhibitor in multiple AML cell lines, patient-derived primary cells, and orthotopic leukemia models. Overall, after starting the study with bioinformatics using gene expression, we showed that the treatment of TAK-981 decreased SUMOylation at the protein level with potent antileukemic effects, resulting in prolonged survival in orthotopic models. Our results should represent sufficient rationale for testing TAK-981 in AML treatment, as it is already being done in clinical trials for solid tumors.
TAK-981 is a highly specific inhibitor of SUMOylation having little effect on ubiquitination or neddylation.20 Still, the mechanism of anticancer activity of TAK-981 may be multifaceted because of the broad-reaching roles of SUMOylation in cancer.5,45 Interestingly, recent data suggest that TAK-981’s activity against solid tumors is dependent on antitumor immunity, especially through type 1 interferon signaling regulated by SUMOylation.16,33 For an immune-competent syngeneic flank model, TAK-981’s activity was abolished when the type 1 interferon receptor was knocked out.33 In addition, in 2 different syngeneic flank models, a survival benefit was observed for the TAK-981–immune checkpoint inhibitor (ICI) combination groups but not for the TAK-981 monotherapy groups, suggesting a cancer cell–extrinsic mechanism of TAK-98133. In our orthotopic models for AML, a hematologic cancer, we observed significant inhibition of leukemia growth and survival benefits in both immune-competent syngeneic mouse transplants and human xenograft models with immune-deficient mice. It should be noted that the NSG immune-deficient mice used here lacked T lymphocytes and had defective dendritic cells that had proved critical to antitumor immunity by TAK-981 in the above solid tumor settings. In addition, we observed potent in vitro inhibitory effects of TAK-981 as well as the induction of differentiation markers for various AML cell lines. Direct apoptotic effects of TAK-981 were also observed ex vivo for primary AML cells from patients. These results strongly suggest that TAK-981 exhibits cancer cell–inherent anti-AML activity. The apparent discrepancy with the above study may be because of the fundamental differences between solid tumors vs AML cancer or the experimental settings (ie, flank transplant vs orthotopic [blood] xenograft).
Still, we do not exclude the possibility of anti-AML immunity by TAK-981 or synergy with ICIs in immune-competent human AML settings that we did not study. For acute leukemia, immunotherapy has been advanced and regularly used in clinics for acute lymphoblastic leukemia, and it has also been rapidly developing for AML,33,48 as evidenced by the approval of gemtuzumab ozogamicin in 2017. At this point, ICI monotherapy for AML has been proven not to be very satisfactory,49,50 and its combinations with hypomethylating agents that have their own immune-modulatory effects51,52 have yielded mixed results.53,54 As for the positive ones, those from a phase 1b study on the combination of azacitidine and magrolimab in patients ineligible for intensive chemotherapy were quite encouraging.53 Notably, this combination was effective even for therapy-refractory patients with TP53-mutated AML, though the overall number of patients was small. Larger human clinical trials with TAK-981-ICI combinations are warranted to evaluate their real effects in human AML.55
We showed that TAK-981 exhibited stronger or similar potency than cytarabine in all the AML cell lines tested as well as in patient-derived primary AML cells. Moreover, TAK-981 exhibited inhibition for cytarabine-resistant AML cell lines in vitro (KG-1 and THP-1 cells; our results and other studies by Bossis et al19 and Ma et al56) as well as in a therapy-resistant in vivo model (MOLM-14 orthotopic xenograft). TAK-981 has also decreased the expression of CD39, whose expression is mediated by SUMOylation.57CD39 has been known to be overexpressed in both cytarabine-resistant AML cells and residual AML cells in patients after chemotherapy.37 Enhancing CD39 expression provoked resistance against cytarabine, whereas inhibiting it improved the response to cytarabine in AML cells.37 These results might explain TAK-981’s strong activity against cells with high IC50 values for cytarabine (>100 nM), such as KG-1, THP-1, and MOLM-14. Considering the different modes of action between TAK-981 and cytarabine and the differences in cell lines and primary cells, it will be interesting to see if their potency difference is maintained in real patient cases. Still, the different mode of action might explain the strong synergy of TAK-981 with current drugs in several settings shown in our study.
It is worthwhile to note that the IC50 values of both TAK-981 and cytarabine for the primary cells were much higher than those for the AML cell lines. With the lower SUMOylation status of primary AML cells than that of the cell lines (supplemental Figure 7) being one explanation, an important consideration is that primary AML cells grow much slower than the established AML cell lines. It is possible that the high IC50 value of TAK-981 in primary AML cells may be because of the lower frequency of cell division. This is clearly the case with cytarabine that it almost completely lost its activity for the primary AML cells, even though it is a standard of care drug. Therefore, the absolute value of the IC50 may not be directly translated into high in vivo toxicity. We believe the much slower proliferation of the primary AML cells should be considered seriously, and, therefore, a correlation analysis between SUMOylation extent and cytotoxicity across primary AML cells and cell lines might not be conclusive.
Overall, the current study provides strong evidence for SUMOylation as a new targetable pathway for AML, based on integrated bioinformatic screening and validations with in vitro, ex vivo, and in vivo preclinical AML models. For toxicity, the longer survival of TAK-981–treated mice indicates a favorable therapeutic index. Consistent with this, a previous study with TAK-981 showed a good toxicity property up to 40 mg/kg in mice.33 In addition, healthy patients or patients with remission after therapy had lower SAE1/SAE2, the target of TAK-981, than patients with active AML (Figure 1E), suggesting possible selectivity of the drug. These favorable efficacy and toxicity data should prompt further studies for its optimal combination and transitions to clinical trials with AML.
Acknowledgments
This work was supported by the Basic Science Research Program through National Research Foundation of Korea grants funded by the Korean Government Ministry of Science and ICT (NRF-2018R1A3B1052328) (S.P.), (MSIT-2022R1A2C2004517) (B.-S.C.), and (NRF-2020R1I1A1A01073124) (J.-M.K.).
Authorship
Contribution: H.S.K., B.-R.K., T.T.P.D., Y.-J.K., H.S., J.-M.K., S.J., D.K., J.K., and Y.J.S. established the methodology, obtained the experimental data, performed the formal analysis, and reviewed the manuscript; J.-M.K. obtained funding, supervised the study, and reviewed the manuscript; H.-J.K. obtained funding, recruited patients, supervised the study, and reviewed the manuscript; and B.-S.C. and S.P. conceptualized the study, obtained funding, supervised the study, and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Byung-Sik Cho; Leukemia Research Institute and Department of Hematology, Catholic Hematology Hospital, Seoul St. Mary's Hospital, The Catholic University of Korea, 222 Banpo-daero, Seocho-gu, Seoul 06591, South Korea; e-mail: cbscho@catholic.ac.kr; and Sunghyouk Park; Natural Products Research Institute, College of Pharmacy, Seoul National University, 1 Gwanak-Ro, Gwanak-gu, Seoul 08826, South Korea; e-mail: psh@snu.ac.kr.

