

Key Points
Fully automated classification of mature B-cell neoplasms based on standardized flow cytometry achieves high specificity.
Algorithm-based results can objectively inform on the necessity of ancillary diagnostic tests during the work-up of these diseases.

Abstract
Reproducible expert-independent flow-cytometric criteria for the differential diagnoses between mature B-cell neoplasms are lacking. We developed an algorithm-driven classification for these lymphomas by flow cytometry and compared it to the WHO gold standard diagnosis. Overall, 662 samples from 662 patients representing 9 disease categories were analyzed at 9 laboratories using the previously published EuroFlow 5-tube-8-color B-cell chronic lymphoproliferative disease antibody panel. Expression levels of all 26 markers from the panel were plotted by B-cell entity to construct a univariate, fully standardized diagnostic reference library. For multivariate data analysis, we subsequently used canonical correlation analysis of 176 training cases to project the multidimensional space of all 26 immunophenotypic parameters into 36 2-dimensional plots for each possible pairwise differential diagnosis. Diagnostic boundaries were fitted according to the distribution of the immunophenotypes of a given differential diagnosis. A diagnostic algorithm based on these projections was developed and subsequently validated using 486 independent cases. Negative predictive values exceeding 92.1% were observed for all disease categories except for follicular lymphoma. Particularly high positive predictive values were returned in chronic lymphocytic leukemia (99.1%), hairy cell leukemia (97.2%), follicular lymphoma (97.2%), and mantle cell lymphoma (95.4%). Burkitt and CD10+ diffuse large B-cell lymphomas were difficult to distinguish by the algorithm. A similar ambiguity was observed between marginal zone, lymphoplasmacytic, and CD10− diffuse large B-cell lymphomas. The specificity of the approach exceeded 98% for all entities. The univariate immunophenotypic library and the multivariate expert-independent diagnostic algorithm might contribute to increased reproducibility of future diagnostics in mature B-cell neoplasms.
Introduction
The 2016 World Health Organization (WHO) classification of lymphoid neoplasms integrates clinical, pathological, immunophenotypic, genetic, and molecular data in order to distinguish between mature B-cell lymphoma entities.1 The precise classification of these entities (also known as B-cell chronic lymphoproliferative diseases [B-CLPD]) has relevant therapeutic implications. Some of these neoplasms are best managed with watchful waiting, whereas aggressive B-cell lymphoma entities in immediate need of therapy constitute the other extreme of the spectrum. Bruton tyrosine kinase inhibitors and other newly introduced targeted therapies have only proven effective for certain types of B-cell lymphomas,2-8 thus further emphasizing the importance of a reliable and reproducible distinction between the entities.
Immunophenotypic data combined with morphology, molecular, genetic, and clinical findings already contribute to the WHO classification of mature B-cell neoplasms. The evaluation of the immunophenotype usually belongs to the initial steps of the work-up of an unknown case and therefore influences which ancillary diagnostic tests are subsequently applied. Compared with other techniques, flow cytometry might be an advantageous method to assess the immunophenotype by virtue of its short turnaround time, its high dynamic range (so that marker density can be evaluated), its high sensitivity for rare cells (facilitating diagnostics using sparsely infiltrated samples), its potential for standardization, and its world-wide availability.9 Despite these advantages, the potential of flow cytometry for classification purposes is currently underutilized10,11 because of 2 principal reasons. Firstly, fully standardized flow cytometry techniques and panels are not available. Secondly, no objective, expert-independent, reproducible flow cytometric diagnostic criteria exist that have been validated in a multicentric setting against the current WHO gold standard diagnosis.
The EuroFlow Consortium had previously developed a 5-tube-8-color antibody panel for the diagnostic classification of mature B-cell neoplasms (B-CLPD panel)9 to be combined with standard operating procedures (SOP) and quality assessment (QA) tools for full technical standardization of the assay.12-15 The antibody panel had been optimized in 7 consecutive developmental rounds aiming at maximum separation between entities and minimal redundancy among the markers.9 Given the careful panel design and the large marker set,10,11,16,17 the panel exhaustively captures relevant immunophenotypic information for the differential diagnosis of mature B-cell neoplasms. However, standardized and objective interpretation of the flow cytometric data generated using this panel has not been demonstrated yet.
In this multicentric study, we designed and validated an expert-independent classification algorithm for mature B-cell neoplasms based on canonical correlation analysis (CCA), which is intended to be used as part of the previously published comprehensive EuroFlow diagnostic work flow.9 Using the novel algorithm, the 26-dimensional flow cytometric data set of the flow cytometric panel can be effectively used for automated classification of these lymphomas and leukemias. We also generated reproducible information on the diagnostic accuracy of the flow cytometric classification results as well as on conceivable differential diagnoses so that ancillary (in particular molecular) diagnostic techniques can be applied depending on the likelihood of a diagnosis and the clinical needs of an individual patient.
Methods
Patients and samples
Between 6 March 2009 and 13 March 2020, 662 samples from an identical number of patients comprising 145 chronic lymphocytic leukemia (CLL), 58 hairy cell leukemia (HCL), 29 Burkitt (BL), 104 diffuse large B-cell (DLBCL), 129 follicular (FL), 74 lymphoplasmacytic (LPL), 76 mantle cell (MCL), and 47 marginal zone (MZL) lymphoma cases were studied (main study, Table 1; details in supplemental Table 1). Details on sample selection are provided in supplemental Methods. In addition, 501 consecutive samples with a suspicion of lymphoma infiltration from 501 patients were acquired at 2 centers between 14 March 2020 and 30 September 2021 in the scope of an independent follow-up study. Institutional review boards of the participating centers approved the pseudonymized usage of leftover sample material for these studies.
Characteristics of patients included into training and validation cohorts
| Parameter | Training cohort (n = 176) | Validation cohort (n = 486) | Total | |
|---|---|---|---|---|
| WHO diagnosis | BL | 16 | 13 | 29 |
| CLL | 20 | 125 | 145 | |
| DLBCL | 40 | 64 | 104 | |
| FL | 20 | 109 | 129 | |
| HCL | 20 | 38 | 58 | |
| LPL | 20 | 54 | 74 | |
| MCL | 20 | 56 | 76 | |
| MZL | 20 | 27 | 47 | |
| Gender | Female/male | 65/111 | 181/305 | 246/416 |
| Age (y) | Median (range) | 67 (2-93) | 65 (3-95) | 66 (2-95) |
| WBC (109/L)* | Median (range) | 8.1 (0.31-296) | 10 (0.8-217) | 9.4 (0.31-296) |
| % lymphoma cells† | Median (range) | 42.8 (0.12-99) | 41.6 (0.14-99) | 41.8 (0.12-99) |
| Disease phase | Diagnosis | 153 | 421 | 574 |
| Follow-up | 10 | 24 | 34 | |
| Relapse | 13 | 41 | 54 | |
| Sample type | PB | 55 | 201 | 256 |
| BM | 65 | 163 | 228 | |
| LN | 41 | 87 | 128 | |
| TM | 8 | 24 | 32 | |
| Other | 7 | 11 | 18 |
| Parameter | Training cohort (n = 176) | Validation cohort (n = 486) | Total | |
|---|---|---|---|---|
| WHO diagnosis | BL | 16 | 13 | 29 |
| CLL | 20 | 125 | 145 | |
| DLBCL | 40 | 64 | 104 | |
| FL | 20 | 109 | 129 | |
| HCL | 20 | 38 | 58 | |
| LPL | 20 | 54 | 74 | |
| MCL | 20 | 56 | 76 | |
| MZL | 20 | 27 | 47 | |
| Gender | Female/male | 65/111 | 181/305 | 246/416 |
| Age (y) | Median (range) | 67 (2-93) | 65 (3-95) | 66 (2-95) |
| WBC (109/L)* | Median (range) | 8.1 (0.31-296) | 10 (0.8-217) | 9.4 (0.31-296) |
| % lymphoma cells† | Median (range) | 42.8 (0.12-99) | 41.6 (0.14-99) | 41.8 (0.12-99) |
| Disease phase | Diagnosis | 153 | 421 | 574 |
| Follow-up | 10 | 24 | 34 | |
| Relapse | 13 | 41 | 54 | |
| Sample type | PB | 55 | 201 | 256 |
| BM | 65 | 163 | 228 | |
| LN | 41 | 87 | 128 | |
| TM | 8 | 24 | 32 | |
| Other | 7 | 11 | 18 |
BM, bone marrow; LN, lymph node; PB, peripheral blood; TM, tumor mass; WBC, white blood count.
Exact WBC is not available for 125 cases.
As percentage of all leukocytes in sample.
Immunophenotypic studies, data analysis, and construction of a diagnostic algorithm for B-cell lymphoma classification
For the main study, samples were stained using a standardized stain-lyse method for cell membrane markers and the EuroFlow B-CLPD panel9 (supplemental Table 2) as described in supplemental Methods and available in regularly updated versions at www.euroflow.org.
The overall data analysis strategy is detailed in Figure 1, supplemental Table 3, and supplemental Methods.
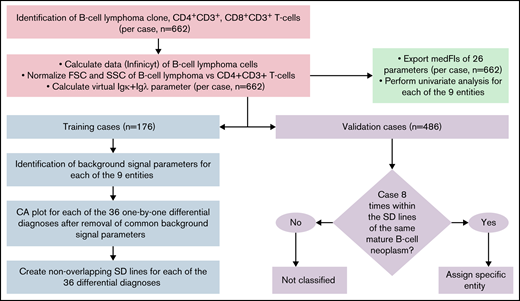
Flowchart of data analysis strategy to create the diagnostic library of expression levels, to develop the diagnostic database, and to validate it. The B-cell lymphoma clone, as well as CD4+CD3+, and CD8+CD3+ T cells were gated individually in each sample. The Calculate Data function of Infinicyt was used to assign each of the 26 parameters of the B-CLPD panel to each malignant B cell in a sample. A virtual immunoglobulin (Ig) κ plus Igλ parameter was created. FSC and SSC of the B-CLPD clone of a sample were normalized vs CD4+CD3+ T cells (red boxes). Subsequently, medFI from all samples were exported and a univariate analysis per entity performed (green box). The data set was divided into training (blue) and validation (purple) sets. In training set cases, parameters with predominantly background signal were identified. Comparative plots of CA1 vs CA2 per differential diagnosis were created using all parameters, but the ones with predominantly background signal for both entities in a given differential diagnosis. Nonoverlapping SD lines were drawn per differential diagnosis using the comparative plot created using the training set cases. If a median of a validation case was included into these SD lines for all 8 possible differential diagnoses of a given entity, that diagnosis was automatically assigned to the sample. CA, canonical axis; FSC, forward scatter; SD, standard deviation; SSC, side scatter.
Flowchart of data analysis strategy to create the diagnostic library of expression levels, to develop the diagnostic database, and to validate it. The B-cell lymphoma clone, as well as CD4+CD3+, and CD8+CD3+ T cells were gated individually in each sample. The Calculate Data function of Infinicyt was used to assign each of the 26 parameters of the B-CLPD panel to each malignant B cell in a sample. A virtual immunoglobulin (Ig) κ plus Igλ parameter was created. FSC and SSC of the B-CLPD clone of a sample were normalized vs CD4+CD3+ T cells (red boxes). Subsequently, medFI from all samples were exported and a univariate analysis per entity performed (green box). The data set was divided into training (blue) and validation (purple) sets. In training set cases, parameters with predominantly background signal were identified. Comparative plots of CA1 vs CA2 per differential diagnosis were created using all parameters, but the ones with predominantly background signal for both entities in a given differential diagnosis. Nonoverlapping SD lines were drawn per differential diagnosis using the comparative plot created using the training set cases. If a median of a validation case was included into these SD lines for all 8 possible differential diagnoses of a given entity, that diagnosis was automatically assigned to the sample. CA, canonical axis; FSC, forward scatter; SD, standard deviation; SSC, side scatter.
Infinicyt software (Cytognos SL, Salamanca, Spain, developmental version 2.0.3 a.B-CLPD_S3) was used to analyze the flow cytometry data. The malignant clone of each of the 662 lymphoma and leukemia cases was electronically separated from the other cellular events via CD45, CD19, CD20, FSC, and SSC to obtain a purity of >90% malignant cells prior to further analysis (supplemental Figure 1). The nearest neighbor algorithm18 as implemented into Infinicyt software was applied to CD20, CD45, CD19, FSC, and SSC so that a value for CD20, CD45, CD19, Igλ, Igκ, CD5, CD38, CD23, CD10, CD79b, CD200, CD43, CD31, CD305, CD11c, IgM, CD81, CD103, CD95, CD22, CD185, CD49d, CD62L, CD39, HLA-DR, and CD27 was assigned to each B-CLPD cell in a sample. The sum of Igκ and Igλ intensities was used instead of the individual immunoglobulin light chains. The resulting virtual Igκ plus Igλ parameter represents the level of immunoglobulin light chain expression but is indifferent to the individual B-cell clones’ restriction toward Igκ or Igλ. Bystander T-cell subpopulations were gated for QA and for normalization of scatter parameters of the clonal B cells. DLBCL cases were divided into CD10+ and CD10− groups (CD10+DLBCL, CD10−DLBCL) based on CD10 medFI on the tumor B cells (cutoff: 200 fluorescence channel units, scaled from 0 to 262 143) in order to account for intradisease heterogeneity of this specific lymphoma type.19 Thus, 662 B-CLPD cases represented 9 different lymphoma entities (Table 1). Flow cytometry data from these cases were merged into a single flow cytometry data file and color coded per diagnostic category. This total cohort of 662 cases was used for univariate analyses.
Sixteen to 20 cases (total n = 176) per disease category were randomly selected as training cohort for the diagnostic algorithm (supplemental Methods).
All 36 possible 1 × 1 pairwise differential diagnoses between the 9 B-CLPD entities were represented by individual 2-dimensional comparative dot plots derived from the training cases. The coefficients for the axes of the dot plots (supplemental Table 4) were obtained using CCA,20 which considered all informative parameters (supplemental Table 9) for each individual differential diagnosis. The x-axis (CA1) provides the combination of immunophenotypic parameters with the greatest separation between 2 given lymphoid neoplasms, whereas the y-axis (CA2) contains the combination with the second greatest separation (Figure 2). The greatest nonoverlapping SD obtained for each 1 × 1 pairwise diagnostic comparison for the training cases (supplemental Table 5) in the comparative CCA plots was used as diagnostic decision boundary for the validation phase. If the median value for all neoplastic B cells, of an unknown case, was included in the diagnostic boundary of an entity from a single 2-dimensional comparative plot, that case fulfilled the diagnostic criterion for this differential diagnosis in favor of this entity. For maximum specificity, unknown cases were required to meet the decision criteria for all 8 projections that included a particular entity. If that requirement was met, a case was automatically classified as this lymphoma; otherwise, that unknown case was considered “not classifiable” (Figure 2).
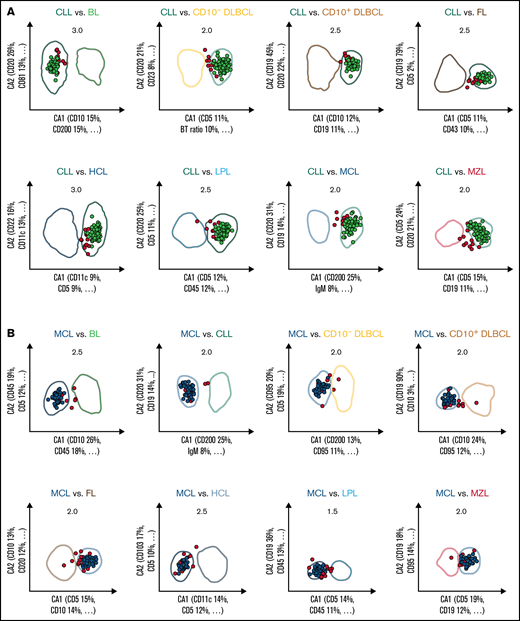
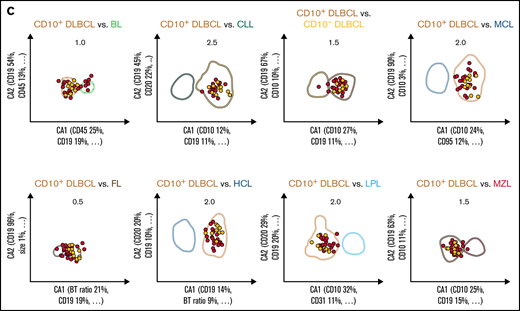
Expert-independent classification of the CLL, MCL, and CD10+ DLBCL validation cohorts. Out of the total possible 36 2-dimensional CCA-based projections, the 8 projections that include CLL (A), MCL (B), and CD10+ DLBCL (C) are shown only. The X- and Y-axes of each plot represent CA1 and CA2. CA1 is the projection that captures most of the information for maximum separation between 2 B-CLPD entities; CA2 is the projection that provides the second-greatest amount of independent information for separation. Numbers in the top part of each plot represent the x fold SD of the immunophenotype shown. Numbers in brackets denote the relative contribution of markers to CA1 and CA2, respectively (see supplemental Table 4 for a full list of markers and coefficients). Each dot represents the median of 1 case from the validation cohort. (A) Cases included into all 8 representations for CLL are shown in green (n = 112); cases not included into all 8 plots for this leukemia are shown in red (“not classified”, n = 13). These 13 cases did not meet all 8 decision criteria for any other lymphoma. (B) Cases included into all 8 representations for MCL are shown in blue (n = 41); cases not included into all 8 plots for that lymphoma are shown in red (n = 14). Thereof, 13 cases did not meet all 8 decision criteria for any other lymphoma, and 1 MCL was misclassified as LPL (data not shown).
Expert-independent classification of the CLL, MCL, and CD10+ DLBCL validation cohorts. Out of the total possible 36 2-dimensional CCA-based projections, the 8 projections that include CLL (A), MCL (B), and CD10+ DLBCL (C) are shown only. The X- and Y-axes of each plot represent CA1 and CA2. CA1 is the projection that captures most of the information for maximum separation between 2 B-CLPD entities; CA2 is the projection that provides the second-greatest amount of independent information for separation. Numbers in the top part of each plot represent the x fold SD of the immunophenotype shown. Numbers in brackets denote the relative contribution of markers to CA1 and CA2, respectively (see supplemental Table 4 for a full list of markers and coefficients). Each dot represents the median of 1 case from the validation cohort. (A) Cases included into all 8 representations for CLL are shown in green (n = 112); cases not included into all 8 plots for this leukemia are shown in red (“not classified”, n = 13). These 13 cases did not meet all 8 decision criteria for any other lymphoma. (B) Cases included into all 8 representations for MCL are shown in blue (n = 41); cases not included into all 8 plots for that lymphoma are shown in red (n = 14). Thereof, 13 cases did not meet all 8 decision criteria for any other lymphoma, and 1 MCL was misclassified as LPL (data not shown).
The consecutive samples included in the follow-up study were stained at least with the first tube of the B-CLPD panel (lymphoid screening tube, supplemental Table 2).
Validation of the diagnostic algorithm
In order to validate the diagnostic algorithm, data from an independent cohort of 486 individual B-cell lymphoma patients from the main study were plotted against the predefined nonoverlapping diagnostic boundaries in the multivariate CCA plots. The diagnostic algorithm was evaluated vs the gold standard WHO diagnosis, which required a predefined minimum set of ancillary diagnostic test results (supplemental Methods for details).
Applicability of the algorithm in consecutive samples
Presence of a B-cell lymphoma, frequency of malignant B cells as proportion of total nucleated cells in the sample, and achievable purity after gating for CD45, CD19, CD20, FSC, and SSC were analyzed using the samples from the follow-up study and the gating strategy described in supplemental Figure 1. The numbers of total contaminating normal B cells in the samples were derived from the events in the gate that expressed the noninvolved light chain. In order to account for the physiological κ/λ ratio of 1.5, the additional contaminating normal B cells expressing the involved light chain were calculated as λ-expressing B cells multiplied with 1.5 and κ-expressing B cells multiplied with 0.67, respectively. The purity of the gate was computed as difference of total cells within the analysis gate minus the total number of nonclonal B cells. The purity was expressed as percentage of total cells in the analysis gate.
Statistical methods
Details on software and statistical tests are provided in supplemental Methods. P < .05 was considered statistically significant.
Results
Comparable fluorescence intensities of T cells irrespective of center, year of acquisition, or sample material
Fluorescence intensities of residual normal T-cell subpopulations identified in the lymphocyte screening tube (LST, equivalent to tube 1 of the B-CLPD panel, supplemental Methods) were calculated to monitor the technical variance caused by staining procedures, instrument set-up, and acquisition for different types of samples in different centers during the duration of the study. Overall, mean coefficient of variation (CV) between all 9 participating centers were numerically higher than intracenter CV for all 5 antigens (CD3, CD5, CD4, CD8, and CD45) but did not reach statistical significance (P > .2, Table 2). Breakdowns of T-cell fluorescence intensities by center, sample material, and year of acquisition did not reveal a significant impact on any of these variables (supplemental Figures 3-5). These data demonstrate that the application of the EuroFlow technical SOP resulted in standardized quantitative antigen expression levels in spite of the participation of 9 different centers, an acquisition period of 12 years, and various sample material sources. Interestingly, the expression levels of the 5 T-cell–associated markers showed similar variance (Table 2) in spite of the fact that different target antigens, using different antibodies and labeled with different fluorochromes, were detected. We can therefore additionally conclude that the variation detected herein likely represents a general feature of flow cytometry according to EuroFlow standards.
Fluorescence intensities of T-cell subpopulations as in-sample quality control
| Antigen and label (population) | MedFI | Intracenter CV | Intercenter CV | P |
|---|---|---|---|---|
| CD4 PacB (CD4+CD3+) | 6 707 | 22.6% | 27.2% | .9 |
| CD3 APC (CD4+CD3+) | 38 786 | 30.2% | 32.8% | 1.0 |
| CD45 PacO (CD4+CD3+) | 5 441 | 21.2% | 25.6% | .6 |
| CD5 PerCP Cy5.5 (CD4+CD3+) | 11 684 | 22.5% | 30.7% | .2 |
| CD8 FITC (CD8+CD3+) | 13 182 | 23.4% | 26.5% | .0 |
| Antigen and label (population) | MedFI | Intracenter CV | Intercenter CV | P |
|---|---|---|---|---|
| CD4 PacB (CD4+CD3+) | 6 707 | 22.6% | 27.2% | .9 |
| CD3 APC (CD4+CD3+) | 38 786 | 30.2% | 32.8% | 1.0 |
| CD45 PacO (CD4+CD3+) | 5 441 | 21.2% | 25.6% | .6 |
| CD5 PerCP Cy5.5 (CD4+CD3+) | 11 684 | 22.5% | 30.7% | .2 |
| CD8 FITC (CD8+CD3+) | 13 182 | 23.4% | 26.5% | .0 |
MedFI of all CD4+CD3+ (n = 650) and CD8+CD3+ (n = 652) populations, mean intracenter CV, and intercenter CV. P values relate to the comparison between intra- and intercenter CV.
APC, allophycocyanin; CV, coefficient of variation; FITC, fluorescein isothiocyanate; PacB, Pacific Blue; PacO, Pacific Orange; PerCP, peridinin-chlorophyll-protein.
Construction of a comprehensive library of flow cytometric marker expression levels by B-cell lymphoma disease category
The large data set of standardized flow cytometric data provided the opportunity to create a comprehensive library of individual B-cell lymphoma–associated marker expression levels per entity. A detailed map for fast visual inspection of differences in expression levels per marker was obtained (Figure 3; supplemental Table 6). In brief, each of the 26 parameters shows significant differences between the entities, but quantitative expression levels commonly overlap between the lymphoma categories. The diagnostic significance of individual markers for particular lymphoma entities are elucidated in supplemental Results. The description of the observed medians and variance of these parameters also facilitate an appreciation as to why particular markers contribute to comparative CCA-based plots that maximize separation between entities (Tables 3 and 4; supplemental Table 4).
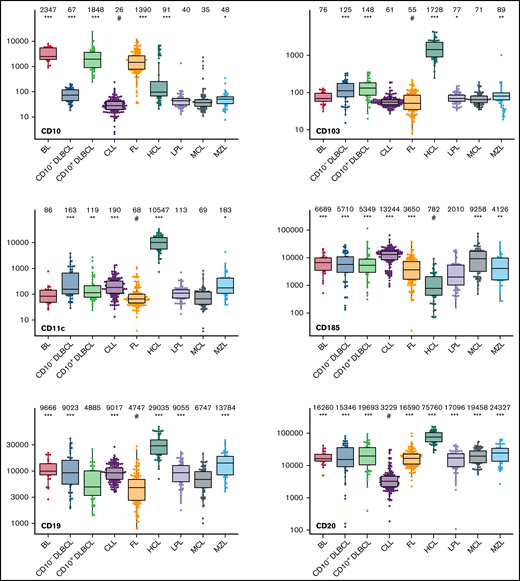
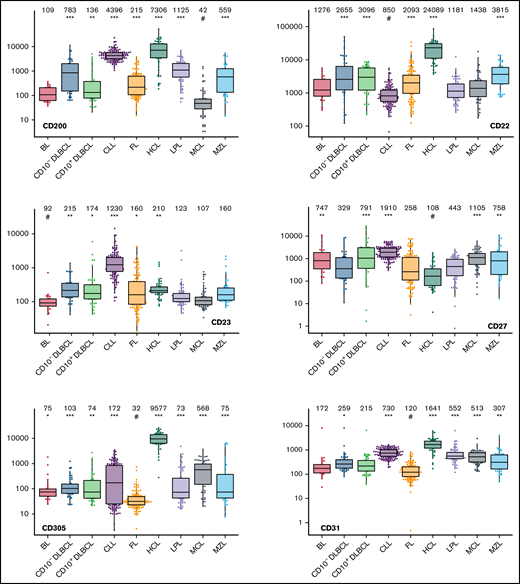
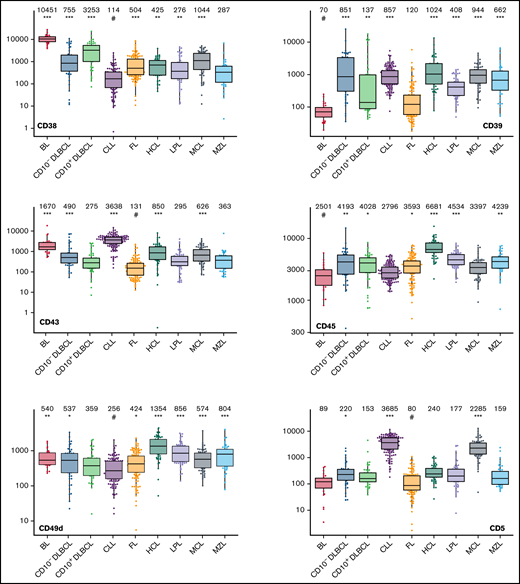
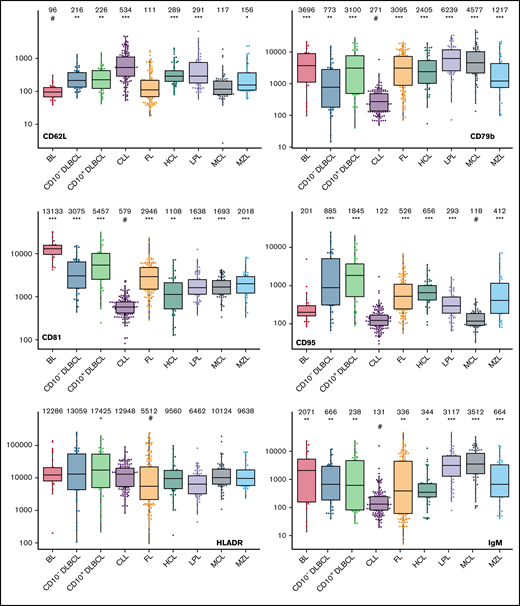
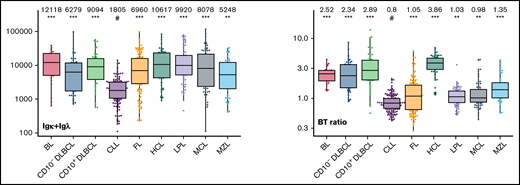
Box plots with univariate representation of the expression levels of all 26 parameters assessed by disease entity. The names of the parameters are shown in the lower right or left corner. Marker expression in log scale. Horizontal lines indicate medians, boxes show interquartile ranges, and whiskers extend to largest/smallest value within the median plus or minus 1.5× interquartile range. Dots show cases out of the interquartile range. Each case is represented by its medFI (n = 662). Median values per marker and entity are specified at the top of each diagram. The symbol “#” indicates the lowest medFI for each marker; significance of higher medFI are indicated as follows: *P < .01; **P < .001; ***P < .0001.
Box plots with univariate representation of the expression levels of all 26 parameters assessed by disease entity. The names of the parameters are shown in the lower right or left corner. Marker expression in log scale. Horizontal lines indicate medians, boxes show interquartile ranges, and whiskers extend to largest/smallest value within the median plus or minus 1.5× interquartile range. Dots show cases out of the interquartile range. Each case is represented by its medFI (n = 662). Median values per marker and entity are specified at the top of each diagram. The symbol “#” indicates the lowest medFI for each marker; significance of higher medFI are indicated as follows: *P < .01; **P < .001; ***P < .0001.
Most discriminatory markers per differential diagnosis
| BL | CD10− DLBCL | CD10+ DLBCL | CLL | FL | HCL | LPL | MCL | |
|---|---|---|---|---|---|---|---|---|
| CD10− DLBCL | CD10 (20.7%) | |||||||
| CD45 (16.6%) | ||||||||
| CD10+ DLBCL | CD45 (24.8%) | CD10 (29.4%) | ||||||
| CD19 (18.5%) | CD19 (11.7%) | |||||||
| CLL | CD10 (15.2%) | CD5 (10.9%) | CD10 (12%) | |||||
| CD200 (14.5%) | BT ratio (10.1%) | CD19 (11.3%) | ||||||
| FL | CD45 (23.3%) | CD10 (20.9%) | BT ratio (19.9%) | CD5 (10.8%) | ||||
| CD38 (14.9%) | CD43 (9.7%) | CD19 (18.2%) | CD43 (9.7%) | |||||
| HCL | CD38 (11.1%) | CD305 (20.7%) | CD19 (14.0%) | CD11c (8.9%) | CD305 (16.4%) | |||
| CD305 (11.0%) | CD103 (13.9%) | CD22 (8.7%) | CD5 (8.8%) | CD11c (14.1%) | ||||
| LPL | CD45 (18.4%) | CD45 (13.0%) | CD10 (21.9%) | CD45 (12.3%) | CD10 (17.6%) | CD45 (16.7%) | ||
| CD10 (17.9%) | CD22 (12.7%) | CD31 (11.2%) | CD5 (11.9%) | CD31 (16.7%) | CD11c (10.8%) | |||
| MCL | CD10 (25.9%) | CD200 (12.5%) | CD10 (24.4%) | CD200 (25.3%) | CD5 (15.4%) | CD11c (14.0%) | CD5 (14.3%) | |
| CD45 (17.8%) | CD95 (10.9%) | CD95 (11.8%) | IgM (8.0%) | CD10 (13.6%) | CD5 (11.8%) | CD45 (10.7%) | ||
| MZL | CD10 (19.2%) | CD20 (9.0%) | CD10 (25.2%) | CD5 (15%) | CD10 (20.8%) | CD45 (18.1%) | CD22 (11.3%) | CD5 (19.4%) |
| CD45 (15.9%) | CD81 (8.9%) | CD19 (15.6%) | CD19 (11.2%) | BT ratio (10.6%) | CD103 (15.2%) | CD31 (8.9%) | CD19 (12.2%) |
| BL | CD10− DLBCL | CD10+ DLBCL | CLL | FL | HCL | LPL | MCL | |
|---|---|---|---|---|---|---|---|---|
| CD10− DLBCL | CD10 (20.7%) | |||||||
| CD45 (16.6%) | ||||||||
| CD10+ DLBCL | CD45 (24.8%) | CD10 (29.4%) | ||||||
| CD19 (18.5%) | CD19 (11.7%) | |||||||
| CLL | CD10 (15.2%) | CD5 (10.9%) | CD10 (12%) | |||||
| CD200 (14.5%) | BT ratio (10.1%) | CD19 (11.3%) | ||||||
| FL | CD45 (23.3%) | CD10 (20.9%) | BT ratio (19.9%) | CD5 (10.8%) | ||||
| CD38 (14.9%) | CD43 (9.7%) | CD19 (18.2%) | CD43 (9.7%) | |||||
| HCL | CD38 (11.1%) | CD305 (20.7%) | CD19 (14.0%) | CD11c (8.9%) | CD305 (16.4%) | |||
| CD305 (11.0%) | CD103 (13.9%) | CD22 (8.7%) | CD5 (8.8%) | CD11c (14.1%) | ||||
| LPL | CD45 (18.4%) | CD45 (13.0%) | CD10 (21.9%) | CD45 (12.3%) | CD10 (17.6%) | CD45 (16.7%) | ||
| CD10 (17.9%) | CD22 (12.7%) | CD31 (11.2%) | CD5 (11.9%) | CD31 (16.7%) | CD11c (10.8%) | |||
| MCL | CD10 (25.9%) | CD200 (12.5%) | CD10 (24.4%) | CD200 (25.3%) | CD5 (15.4%) | CD11c (14.0%) | CD5 (14.3%) | |
| CD45 (17.8%) | CD95 (10.9%) | CD95 (11.8%) | IgM (8.0%) | CD10 (13.6%) | CD5 (11.8%) | CD45 (10.7%) | ||
| MZL | CD10 (19.2%) | CD20 (9.0%) | CD10 (25.2%) | CD5 (15%) | CD10 (20.8%) | CD45 (18.1%) | CD22 (11.3%) | CD5 (19.4%) |
| CD45 (15.9%) | CD81 (8.9%) | CD19 (15.6%) | CD19 (11.2%) | BT ratio (10.6%) | CD103 (15.2%) | CD31 (8.9%) | CD19 (12.2%) |
Each cell of the table contains the marker with highest weight contributing to the first canonical axis on top and the marker with the second-highest contribution at the bottom. The significance of the markers is provided in brackets.
Contribution of individual markers to multivariate differential diagnosis
Given the overlap in expression levels of individual markers between the entities, a univariate analysis of the data set cannot provide a straightforward diagnostic classification of individual patients. In order to both minimize intra-disease variation and maximize distances within the 26-dimensional data space, we therefore applied CCA for multivariate analysis and dimension reduction. The method provided the combination of flow cytometric markers resulting in the greatest separation between any 2 lymphoma entities (Table 3; supplemental Table 4). As expected, the relevance of individual markers differed by pairwise differential diagnosis. A total of 17 markers contributed with the highest or second-highest weight toward any of the 36 first canonical axes we obtained from the whole data set (Table 3). The marker that contributed most often with high relevance was CD10. The restriction to the 2 most relevant markers provides a useful orientation on the relative importance of markers in a multivariate analysis. However, it should be noted that for certain differential diagnoses, the separation in the discriminatory plots decreased substantially when markers with lesser weight were electronically removed (data not shown).
Validation of the automated classification algorithm for mature B-cell neoplasms
We tested the diagnostic performance of the approach for unequivocal classification of mature B-cell neoplasms using an independent cohort of validation cases. We detected differences by disease category (Table 4).
Algorithm-based flow cytometric diagnosis (486 validation cases)
| WHO diagnosis | n | Algorithm-based flow cytometric diagnosis | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BL | CD10− DLBCL | CD10+ DLBCL | CLL | FL | HCL | LPL | MCL | MZL | NC | Sensitivity | Specificity | PPV | NPV | |||
| T1 to T5 | BL | 13 | 7 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 6 | 54.1 ± 13.8% | 98.9 ± 0.5% | 59.3 ± 12.4% | 98.7 ± 0.4% |
| CD10− DLBCL | 31 | 0 | 5 | 0 | 0 | 0 | 0 | 0 | 0 | 5 | 21 | 16.2 ± 6.6% | 99.3 ± 0.4% | 63.4 ± 18.1% | 94.6 ± 0.4% | |
| CD10+ DLBCL | 33 | 4 | 0 | 7 | 0 | 1 | 0 | 0 | 0 | 0 | 21 | 21.0 ± 6.9% | 98.2 ± 0.6% | 46.8 ± 12.8% | 94.5 ± 0.5% | |
| CLL | 125 | 0 | 0 | 0 | 112 | 0 | 0 | 0 | 0 | 0 | 13 | 89.7 ± 2.7% | 99.7 ± 0.3% | 99.1 ± 0.9% | 96.6 ± 0.9% | |
| FL | 109 | 1 | 0 | 8 | 0 | 34 | 0 | 1 | 0 | 0 | 65 | 31.3 ± 4.5% | 99.7 ± 0.2% | 97.2 ± 2.6% | 83.4 ± 0.9% | |
| HCL | 38 | 0 | 0 | 0 | 0 | 0 | 34 | 0 | 0 | 0 | 4 | 89.4 ± 5.2% | 99.8 ± 0.2% | 97.2 ± 2.6% | 99.1 ± 0.4% | |
| LPL | 54 | 0 | 2 | 0 | 0 | 0 | 0 | 17 | 1 | 2 | 32 | 31.4 ± 6.1% | 99.5 ± 0.3% | 89.2 ± 6.9% | 92.1 ± 0.6% | |
| MCL | 56 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 41 | 0 | 14 | 73.2 ± 5.8% | 99.5 ± 0.3% | 95.4 ± 3.1% | 96.6 ± 0.7% | |
| MZL | 27 | 0 | 1 | 0 | 1 | 0 | 1 | 0 | 1 | 7 | 16 | 26.4 ± 8.4% | 98.5 ± 0.5% | 50.7 ± 12.2% | 95.8 ± 0.5% | |
| T1 + T2 | BL | 13 | 7 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 6 | 53.8 ± 13.5% | 97.7 ± 0.7% | 39.4 ± 9.5% | 98.7 ± 0.4% |
| CD10− DLBCL | 31 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 28 | 6.7 ± 4.5% | 99.6 ± 0.3% | 50.9 ± 28.6% | 94.0 ± 0.3% | |
| CD10+ DLBCL | 33 | 7 | 0 | 8 | 0 | 1 | 1 | 0 | 0 | 0 | 16 | 24.7 ± 7.4% | 97.6 ± 0.7% | 42.8 ± 10.1% | 94.7 ± 0.5% | |
| CLL | 125 | 0 | 0 | 0 | 108 | 0 | 0 | 0 | 0 | 1 | 16 | 86.3 ± 3.0% | 99.7 ± 0.3% | 99.1 ± 0.9% | 95.5 ± 0.9% | |
| FL | 109 | 4 | 1 | 10 | 0 | 37 | 0 | 0 | 0 | 1 | 56 | 33.8 ± 4.4% | 99.5 ± 0.4% | 95.1 ± 3.3% | 83.9 ± 0.9% | |
| HCL | 38 | 0 | 0 | 0 | 0 | 0 | 11 | 0 | 0 | 1 | 26 | 28.8 ± 7.4% | 99.8 ± 0.2% | 91.4 ± 8.3% | 94.3 ± 0.6% | |
| LPL | 54 | 0 | 1 | 0 | 0 | 1 | 0 | 8 | 1 | 1 | 42 | 15.0 ± 4.8% | 99.5 ± 0.3% | 80.4 ± 12.9% | 90.4 ± 0.5% | |
| MCL | 56 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 26 | 0 | 28 | 46.3 ± 6.8% | 99.5 ± 0.3% | 92.8 ± 4.8% | 93.4 ± 0.8% | |
| MZL | 27 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 1 | 1 | 23 | 3.6 ± 3.5% | 98.9 ± 0.5% | 17.3 ± 17.9% | 94.6 ± 0.2% | |
| WHO diagnosis | n | Algorithm-based flow cytometric diagnosis | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BL | CD10− DLBCL | CD10+ DLBCL | CLL | FL | HCL | LPL | MCL | MZL | NC | Sensitivity | Specificity | PPV | NPV | |||
| T1 to T5 | BL | 13 | 7 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 6 | 54.1 ± 13.8% | 98.9 ± 0.5% | 59.3 ± 12.4% | 98.7 ± 0.4% |
| CD10− DLBCL | 31 | 0 | 5 | 0 | 0 | 0 | 0 | 0 | 0 | 5 | 21 | 16.2 ± 6.6% | 99.3 ± 0.4% | 63.4 ± 18.1% | 94.6 ± 0.4% | |
| CD10+ DLBCL | 33 | 4 | 0 | 7 | 0 | 1 | 0 | 0 | 0 | 0 | 21 | 21.0 ± 6.9% | 98.2 ± 0.6% | 46.8 ± 12.8% | 94.5 ± 0.5% | |
| CLL | 125 | 0 | 0 | 0 | 112 | 0 | 0 | 0 | 0 | 0 | 13 | 89.7 ± 2.7% | 99.7 ± 0.3% | 99.1 ± 0.9% | 96.6 ± 0.9% | |
| FL | 109 | 1 | 0 | 8 | 0 | 34 | 0 | 1 | 0 | 0 | 65 | 31.3 ± 4.5% | 99.7 ± 0.2% | 97.2 ± 2.6% | 83.4 ± 0.9% | |
| HCL | 38 | 0 | 0 | 0 | 0 | 0 | 34 | 0 | 0 | 0 | 4 | 89.4 ± 5.2% | 99.8 ± 0.2% | 97.2 ± 2.6% | 99.1 ± 0.4% | |
| LPL | 54 | 0 | 2 | 0 | 0 | 0 | 0 | 17 | 1 | 2 | 32 | 31.4 ± 6.1% | 99.5 ± 0.3% | 89.2 ± 6.9% | 92.1 ± 0.6% | |
| MCL | 56 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 41 | 0 | 14 | 73.2 ± 5.8% | 99.5 ± 0.3% | 95.4 ± 3.1% | 96.6 ± 0.7% | |
| MZL | 27 | 0 | 1 | 0 | 1 | 0 | 1 | 0 | 1 | 7 | 16 | 26.4 ± 8.4% | 98.5 ± 0.5% | 50.7 ± 12.2% | 95.8 ± 0.5% | |
| T1 + T2 | BL | 13 | 7 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 6 | 53.8 ± 13.5% | 97.7 ± 0.7% | 39.4 ± 9.5% | 98.7 ± 0.4% |
| CD10− DLBCL | 31 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 28 | 6.7 ± 4.5% | 99.6 ± 0.3% | 50.9 ± 28.6% | 94.0 ± 0.3% | |
| CD10+ DLBCL | 33 | 7 | 0 | 8 | 0 | 1 | 1 | 0 | 0 | 0 | 16 | 24.7 ± 7.4% | 97.6 ± 0.7% | 42.8 ± 10.1% | 94.7 ± 0.5% | |
| CLL | 125 | 0 | 0 | 0 | 108 | 0 | 0 | 0 | 0 | 1 | 16 | 86.3 ± 3.0% | 99.7 ± 0.3% | 99.1 ± 0.9% | 95.5 ± 0.9% | |
| FL | 109 | 4 | 1 | 10 | 0 | 37 | 0 | 0 | 0 | 1 | 56 | 33.8 ± 4.4% | 99.5 ± 0.4% | 95.1 ± 3.3% | 83.9 ± 0.9% | |
| HCL | 38 | 0 | 0 | 0 | 0 | 0 | 11 | 0 | 0 | 1 | 26 | 28.8 ± 7.4% | 99.8 ± 0.2% | 91.4 ± 8.3% | 94.3 ± 0.6% | |
| LPL | 54 | 0 | 1 | 0 | 0 | 1 | 0 | 8 | 1 | 1 | 42 | 15.0 ± 4.8% | 99.5 ± 0.3% | 80.4 ± 12.9% | 90.4 ± 0.5% | |
| MCL | 56 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 26 | 0 | 28 | 46.3 ± 6.8% | 99.5 ± 0.3% | 92.8 ± 4.8% | 93.4 ± 0.8% | |
| MZL | 27 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 1 | 1 | 23 | 3.6 ± 3.5% | 98.9 ± 0.5% | 17.3 ± 17.9% | 94.6 ± 0.2% | |
The upper half of the table describes the results when tubes 1 to 5 of the B-CLPD panel are used; the bottom half tabulates the results from the same validation set cases using tubes 1 and 2 of the B-CLPD panel only. Mean plus SD of sensitivity, specificity, PPV, and NPV were calculated by bootstrapping.
NC, not classified, NPV, negative predictive value; PPV, positive predictive value.
Out of a total of 486 validation cases, the algorithm offered the diagnosis of CLL for 113 samples. This diagnosis was 112 times correct, with 1 case classified as CLL by the automated algorithm but as MZL by WHO criteria (PPV 99.1%). For 373 validation cases, the algorithm either assigned another lymphoma or was unable to classify, thus yielding a diagnosis of not being a CLL. Most cases without a CLL diagnosis assigned by the algorithm (360/373 cases) corresponded, indeed, to an entity different from CLL, whereas 13 CLL cases (all assigned to the “not classified” category) were not selected by the algorithm as CLL (NPV 96.6%). It follows that every time the algorithm assigns a case to another lymphoma category (and not to “unclassifiable”), a diagnosis of CLL is unlikely. In contrast, whenever the diagnosis of CLL is automatically made, this is very likely correct.
Similarly, of the 35 times a HCL was diagnosed by the algorithm, agreement with the WHO-based diagnosis was obtained in 34 cases, whereas 1 MZL as per WHO criteria was misdiagnosed as HCL (PPV 97.2%). For 451 validation cases, the algorithm either assigned another B-cell lymphoma or was unable to classify, thus yielding a diagnosis of not being a HCL. Only 4 HCL cases in the validation cohort (all assigned to “not classifiable”) were missed (NPV 99.1%). A HCL is therefore an unlikely diagnosis unless that diagnosis is made or the case is at least assigned to the “not classifiable” category.
A similarly high PPV was also found for both MCL (95.4%, 1 LPL, and 1 MZL wrongly classified as MCL by the automated algorithm) and FL (97.2%, 1 CD10+DLBCL misclassified as FL). This was accompanied by a very good NPV in MCL (only 1 case assigned to LPL, all other missed cases assigned to the “not classifiable” category) and a moderate NPV in FL (65 “not classifiable”, 8 recognized as DLBCL, 1 as BL, and 1 as LPL). The algorithm frequently assigned BL cases by WHO to the CD10+DLBCL category (PPV 59.3%).
A diagnosis of LPL was correct in the great majority of cases but was also made in 1 case of FL and 1 MCL (PPV 89.2%). An MZL diagnosis was also seen in CD10−DLBCL and LPL (PPV 50.7%). This translates into an immunophenotypic overlap of CD10−DLBCL (PPV 63.4%) vs LPL and MZL, as well as of CD10+DLBCL (PPV 46.8%) vs FL.
We also explored a possible impact of sample material on the accuracy of the proposed diagnostic algorithm. No FL was misclassified in PB, and there was 1 misclassification in BM, whereas 8 FL cases were wrongly classified (7 CD10+ DLBCL, 1 BL) when lymph node material was investigated by flow cytometry (P = .004). It follows that the NPV for FL is high unless lymph node material is acquired. There was no detectable correlation between sample material and classification results of the automated algorithm in BL, CD10+DLBCL, CD10−DLBCL, CLL, HCL, LPL, MCL, or MZL (P > .3 for each of the entities).
False positive samples for any disease category were scarce, making a high specificity of at least 98% a key feature of the algorithm. In turn, the sensitivity of the test was high in CLL (89.7%), HCL (89.4%), and MCL (73.2%), whereas the entity was often not unequivocally classified in DLBCL, FL, LPL, BL, and MZL (Table 4).
We additionally demonstrated by Monte Carlo cross-validation that the random cohort of training cases used to establish the algorithm preserved the structure of the total data set (supplemental Table 7). Different training cohorts would have yielded similar results by automated analysis.
A stepwise diagnostic approach using data from tubes 1 and 2 of the EuroFlow B-CLPD panel only
In order to facilitate a more cost-effective usage of consumables, we additionally tested a diagnostic approach that relied upon information from tubes 1 and 2 only (Table 4; Figure 4; supplemental Figure 2). This reduced approach correctly detected 108 CLL cases (instead of 112 when all 5 tubes from the B-CLPD panel were used) and, as with the approach using the complete panel, misdiagnosed 1 single MZL (PPV 99.1%). Very similar to the performance of the full panel, 37 FL cases could be correctly diagnosed, whereas 1 CD10+DLBCL and 1 LPL case were erroneously assigned to the FL category (PPV 95.1%). For all other disease categories, the omission of tubes 3 to 5 of the EuroFlow B-CLPD panel resulted in a significant increase in cases assigned to the “not classified” category and/or a drop in PPV. MCL cases were correctly classified by the algorithm with similar PPV when tubes 1 and 2 only or the full panel were used. However, many more patients were assigned to “not classifiable” using the reduced approach. We conclude that for cases with a high pretest likelihood of CLL, FL, or MCL staining of tube 1 and 2 suffices as a first step. Cases that are subsequently, indeed, diagnosed as CLL, FL, or MCL using the reduced panel information do not require additional tests, as flow cytometry data from tubes 3 to 5 will not provide significant additional diagnostic information. Cases without such a diagnosis will require tubes 3 to 5 of the B-CLPD panel to be added in a second step (Figure 4).
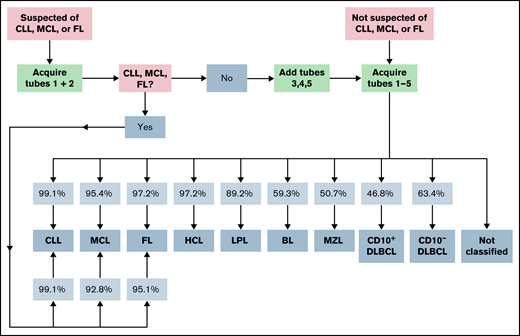
Proposed diagnostic strategy using the expert-independent classification and modular design of the B-CLPD panel. The infiltration of sample by a B-CLPD is assessed using tube 1 first. Patients presenting with a high pretest (clinical, basic laboratory, cytology, or after evaluating tube 1) likelihood for a CLL, MCL, or FL should be evaluated as next step using tubes 1 and 2 only. If the diagnosis of 1 of those 3 entities is made, no further testing is recommended. If neither CLL, nor MCL, nor FL is diagnosed, the additional evaluation of tubes 3 to 5 is advisable. Patients who are not suspected of CLL, MCL, or FL should be directly tested with the full panel. Percentages in squares reflect PPV by entity and testing strategy.
Proposed diagnostic strategy using the expert-independent classification and modular design of the B-CLPD panel. The infiltration of sample by a B-CLPD is assessed using tube 1 first. Patients presenting with a high pretest (clinical, basic laboratory, cytology, or after evaluating tube 1) likelihood for a CLL, MCL, or FL should be evaluated as next step using tubes 1 and 2 only. If the diagnosis of 1 of those 3 entities is made, no further testing is recommended. If neither CLL, nor MCL, nor FL is diagnosed, the additional evaluation of tubes 3 to 5 is advisable. Patients who are not suspected of CLL, MCL, or FL should be directly tested with the full panel. Percentages in squares reflect PPV by entity and testing strategy.
Assessment of the applicability of the diagnostic approach
We analyzed detection rates and numbers of samples with a B-cell clone purity of at least 90% in 501 consecutive samples from the follow-up study. Two hundred and eight samples (42%) contained a B-cell lymphoma that represented between 0.019% and 94.7% of all nucleated cells of that sample. Twenty-eight of those 208 samples with detectable infiltration (13%) exhibited more than 10% benign B-cell contamination of the analysis gate, whereas the contamination with benign B-cell was less than 90% in 87% (180/208) of all infiltrated samples. The median infiltration was significantly lower in samples that could not be purified (1.6%) compared with those in which we achieved the required purity using backbone marker gating (32.1%, P < .0001).
Discussion
Here we show that application of the EuroFlow B-CLPD antibody panel (with 26 parameters) provides a comprehensive, fully reproducible library of antigen expression levels and scatter parameters for the 9 major B-cell lymphoma categories, assessed in 9 centers in a representative cohort of cases. An independent diagnosis based on histology, cytology, molecular biology, and immunophenotype was available for all 662 patients included in the study.
We herein developed a multivariate strategy to effectively use the 26-dimensional comprehensive set of fully standardized flow cytometric data for an unequivocal diagnosis in individual patients. CCA allowed the identification of the most valuable set of markers for a particular differential diagnosis so that optimized 2-dimensional discrimination plots could be constructed. Moreover, based on the training set cases, the acceptance criteria were fitted to the separation of a particular pair of entities, resulting in nonoverlapping boundaries. Finally, the diagnostic algorithm required that identical results are obtained in all 8 possible differential diagnoses for each disease category. In summary, the overall diagnostic strategy was designed to achieve maximum specificity.
By definition, the algorithm excludes that more than 1 diagnosis is made in each case. In contrast to other algorithms,17,21 this rigid approach resulted in a sizeable number of cases that are not assigned to any particular lymphoma entity, thus reducing sensitivity. However, we feel that the inclusion into the “not classified” category is of great clinical utility as it informs of the necessity for ancillary testing by histology, cytogenetics, and/or molecular biology approaches. Although the NPV, except for FL cases tested in lymph node samples, always exceeded 90%, we found particularly high PPVs (>95%) for CLL, HCL, FL, and MCL. This accuracy equals previous single-center observations for CLL and HCL but exceeds published data on FL and MCL.17,21 High PPVs in CLL, HCL, FL, and MCL render targeted molecular confirmatory testing (eg, BRAF V600E mutation in HCL, t[11;14] in MCL) possible, thus facilitating faster and more effective diagnostics. For example, the diagnostic approach described herein allows for a reliable differential diagnosis between MCL and CLL in virtually every case. As many laboratories will start the diagnostic work-up of suspected B-cell lymphoma cases with cytology and flow cytometry, t(11;14) determination by fluorescence in situ hybridization as a second step can be safely omitted in all samples that are automatically assigned to the CLL category by the flow cytometric algorithm.
As expected, high-count monoclonal B lymphocytosis cases were diagnosed as CLL by our approach (data not shown). We recommend determining this differential diagnosis using the absolute counts of malignant B cells in blood for every case automatically assigned to the CLL category.22 A particular challenge for the model is the inclusion of DLBCL cases that were missing in a previous publication.17 CD10+ DLBCL interfered with the correct classification of a fraction of FL and BL cases, whereas a subgroup of CD10− DLBCL cases were difficult to distinguish from LPL and MZL exclusively using phenotypic data (Table 4). Not surprisingly, DLBCL therefore belonged to the disease categories with the lowest PPV and NPV. Nevertheless, we feel that classification models that do not consider this frequent lymphoma that requires distinct treatment would limit the clinical utility of any diagnostic algorithm. A major advantage of this method when compared with previous studies is that, for the first time, diagnostic accuracy of an automated algorithm was based on quantitative flow cytometry data from multiple centers, whereas recently published manuscripts17,21 described single-center investigations.
Positive and negative predictive values are dependent on disease prevalence. Our validation cohort is biased toward rarer disease categories because samples were collected at referral centers and therefore comprises more DLBCL, LPL, MZL, and MCL cases than expected from the prevalence of these lymphomas. In turn, the proportion of CLL cases in our study is lower than the relative prevalence of this leukemia among the B-cell lymphomas as a whole. As a consequence, routine laboratories with a sample accrual that resembles more closely the population prevalence of B-cell lymphoma entities will observe lower PPV for the overrepresented disease categories than stated in Table 4.
Although rarely seen, a few cases were misdiagnosed with the proposed algorithm. In this regard, it should be noted that our gold standard relied on local pathology for confirmatory diagnostics according to WHO. Thus, the multicentric nature of the study comprising 9 centers in different European countries might introduce some variability regarding the WHO gold standard pathology diagnosis. Discordance rates of up to 27.3% have been reported in lymphoma diagnosis between different pathologists.23 In fact, it might be possible that some cases, assigned to a seemingly wrong disease category by the algorithm, were correctly diagnosed whereas the case was locally assigned erroneously to an incorrect WHO group. For instance, case #579 was diagnosed as splenic MZL based upon cytology and histology locally, but BRAF testing was not performed. Flow cytometry showed fluorescence 398 expression levels for CD103 (1207), CD22 (13285), CD305 (6517), and CD11c (4561) all inside the 90th percentile for HCL and outside the 90th percentile for MZL. This case represents the only one falsely diagnosed as HCL by the algorithm. We speculate that with the release of the diagnostic algorithm to the field, such cases in the future might be specifically tested for BRAF V600E mutations and carefully revised by local and reference pathologists.
Cost-effective implementation of the B-CLPD antibody panel is supported by its modular design and integration into the EuroFlow diagnostic workflow.9 At variance to single-step approaches aiming at simultaneous detection and characterization of B-cell non-Hodgkin lymphoma,17 we propose a sequential approach. In a first step, samples will be tested for the presence of a mature B-cell malignancy using the EuroFlow lymphoid screening tube24 (identical to tube 1 of the B-CLPD panel, supplemental Table 2). Whenever a population of clonal B cells is detected in a sample, the B-CLPD panel should be subsequently applied for classification. For normal and reactive samples and those that are infiltrated by a T- or natural killer–cell lymphoma, the full B-CLPD panel will not be applied. Moreover, samples from patients presenting with a suspicion of CLL, MCL, or FL can be stained first with tubes 1 (LST) and 2 only (or only tube 2 is added in case tube 1 has already been stained before). If the algorithm confirms the tentative diagnosis based upon the information from tubes 1 and 2, no further testing is required. If the diagnosis is not confirmed, the additional tubes 3 to 5 should be applied (Figure 4). This modular approach renders possible substantial savings in consumables, time, and human resources because CLL and FL belong to the most frequent diseases in many flow cytometry laboratories.17
We recommend laboratories to apply the B-CLPD panel or at least tubes 1 and 2 thereof for optimal diagnostic performance. However, the univariate flow cytometric data provided as diagnostic library in this manuscript might be also used on a per marker basis. The library includes fully reproducible ranges of observed expression levels for 26 B-cell lymphoma–associated markers. Therefore, the dataset (Figure 3; supplemental Table 6) may be considered a standardized, world-wide applicable reference for future flow cytometry diagnostics25 in this group of diseases. In contrast to the great majority of previous reports on the utility of markers for the differential diagnosis in mature B-cell neoplasms10,11,16,21,26 which used descriptive terms for marker expression profiles, such as “dim” and/or “positivity”, we establish quantitative measures for every individual parameter, provided the EuroFlow SOP and B-CLPD panel reagents are used. Our results can be implemented for routine diagnostics, even when used on a per marker basis only, because EuroFlow offers publicly available QA rounds as a means for external quality control of the staining levels. These same QA rounds also guaranteed technical reproducibility across different centers, and thereby accuracy of the data published in this manuscript.
The multicentric setting of this study, its long duration, and the clearly predefined eligibility criteria, in combination with previously published EuroFlow technical standardization12,13,15,25 and QA,14 will enhance the ability of other laboratories to apply and reproduce the data of this manuscript. We established that the 90% purity criterion for the malignant clone after back-bone marker gating, one of the key inclusion criteria, is attainable in 87% of all infiltrated samples. Expectedly, it was more likely to obtain a highly purified lymphoma population in more heavily infiltrated samples. We predict that adding additional gating markers to the back-bone in future >8-color versions of the B-CLPD panel will increase the percentage of cases that can be sufficiently purified.
The EuroFlow Consortium plans to continuously increase the number of well-annotated cases in the database and will provide updates. We anticipate that up-coming versions will subdivide many of the current 9 disease categories into additional subcategories according to biological and molecular data. For example, at present, both classical HCL and the HCL-variant are classified as HCL. However, a preliminary analysis using CCA revealed that HCL patients without BRAF V600E mutations (typical for HCL-variant27 ) can be completely separated from cases harboring the mutation (typical for classical HCL27 ), mainly driven by CD31, and CD200 (supplemental Figure 8). Once these preliminary observations are confirmed with more cases, both classical HCL and the HCL-variant will most likely form separate, specific categories in the database. We expect this to reduce the overall number of HCL cases in the “not classifiable” group as both subgroups will each occupy a particular, more closely fitting region in the 26-dimensional space created by the immunophenotypic information from the B-CLPD panel. At present, classical HCL and the HCL-variant can be provisionally separated using expression levels: The HCL-variant cases always exhibited a CD200 medFI below 2000 together with a CD31 medFI below 1000, whereas none of the classical HCL cases simultaneously met both features (preliminary data, not shown). Furthermore, up-coming versions of the database and algorithm will also provide flexibility to use the tools described herein on cytometers capable of detecting more than 8 colors.
In summary, algorithm-guided expert-independent flow cytometric classification of mature B-cell neoplasms can contribute to a reliable diagnosis in the majority of these lymphomas. The method will very likely reduce intercenter variation at this particular diagnostic step and will objectively inform on the relative contribution of flow cytometric data to the integrated overall diagnosis of lymphoma entities. For easier worldwide implementation of the newly developed tool by clinical laboratories, the algorithm and database will be publicly released via the Infinicyt software in 2021.
Acknowledgments
The authors acknowledge the valuable scientific contributions of Andy Rawstron, Ruth de Tute, Paulo Lucio, Andrea Mendonca, Vahid Asnafi, Ludovic Lhermitte, Monika Brüggemann, and Juan Perez to the development of the EuroFlow B-CLPD panel. The authors would like to thank Elke Harbst, Linda Falck, Daniel Paape, Beate Wiebeck, Jacqueline Kreitsch, and Patrick Brennan for excellent technical support. Christoph Machka is thanked for critical reading of the manuscript. The authors are grateful to Patrick Brennan for English language proofreading and to Henrik Rudolf for advice on statistical methods.
This work was supported by the EuroFlow Consortium, which received support from the FP6-2004-LIFESCIHEALTH-5 program of the European Commission (grant LSHB-CT-2006-018708) as Specific Targeted Research Project (STREP). The EuroFlow Consortium is part of the European Scientific Foundation for Hemato-Oncology (ESLHO), a Scientific Working Group (SWG) of the European Hematology Association (EHA).
Authorship
Contribution: S.B., J.J.M.v.D., C.E.P., and A.O. designed the research; S.B., R.E., J.J.M.v.D., and A.O. wrote the paper; S.B., R.E., G.G., P.F., J.C., J.F.-M., V.H.J.v.d.V., M.N., J.P., M.R., L.B., and S.L. performed the research; and S.B., R.E., G.G., Q.L., T.K., J.V.V., R.F.R., C.E.P., and A.O. analyzed the data.
Conflict-of-interest disclosure: S.B., C.E.P., J.F.-M., V.H.J.v.d.V., J.J.M.v.D., and A.O. each report being one of the inventors on the EuroFlow-owned patent PCT/NL2010/050332 (methods, reagents, and kits for flow cytometric immunophenotyping of normal, reactive, and malignant leukocytes). The Infinicyt software is based on intellectual property of some EuroFlow laboratories (University of Salamanca, Spain) and the scientific input of other EuroFlow members. All above mentioned intellectual property and related patents are licensed to Cytognos (Salamanca, ES) and BD Biosciences (San Jose, CA), which companies pay royalties to the EuroFlow Consortium. These royalties are exclusively used for continuation of the EuroFlow collaboration and sustainability of the EuroFlow Consortium. J.J.M.v.D. and A.O. report an educational services agreement from BD Biosciences (San Jose, CA) and a scientific advisor agreement with Cytognos; all related fees and honoraria are for the involved university departments at Leiden University Medical Center and University of Salamanca. G.G., R.F.R., and J.V.V. are employees of Cytognos. The remaining authors declare no competing financial interests.
A complete list of additional members of the EuroFlow Consortium appears in “ Appendix.”
Correspondence: Sebastian Böttcher, Clinic III (Hematology, Oncology and Palliative Medicine), Special Hematology Laboratory, Rostock University Medical School, Ernst-Heydemann-Strasse 6, 18057 Rostock, Germany; e-mail: sebastian.boettcher@med.uni-rostock.de; and Jacques J. M. van Dongen, Department of Immunology, Leiden University Medical Center (LUMC), Albinusdreef 2, 2333 ZA Leiden, The Netherlands (Building 1, P3-30) P.O. Box 9600, 2300 RC Leiden, The Netherlands; e-mail: J.J.M.van_dongen@LUMC.nl.
Appendix: Additional EuroFlow Consortium participants
Department of Immunohematology and Blood Transfusion, Leiden University Medical Center, Leiden, The Netherlands – W. M. Bitter, B. R. Lubbers, N. J. M. Kleven, C. I. Teodosio, M. Zlei, A. J. van der Sluijs-Gelling, and F. de Bie.
Department of Pediatrics, Leiden University Medical Center, Leiden, The Netherlands – M. van der Burg, M. W. Schilham, and M. van Ostaije-ten Dam.
Department of Immunology, Erasmus MC, Rotterdam, The Netherlands – A. W. Langerak, J. te Marvelde, and J. Schilperoord-Vermeulen.
Department of Medicine, Cancer Research Center, University of Salamanca, Spain – J. Almeida, M. B. Vidriales, M. Pérez-Andrés, S. Matarraz, L. Martín, and N. Puig.
Hemato Oncology Laboratory, Instituto Portugués de Oncologia, Lisbon, Portugal – M. Gomes da Silva, J. Desterro, and T. Faria.
2nd Department of Medicine, University of Schleswig-Holstein – Campus Kiel, Kiel, Germany – M. Brüggemann, M. Szczepanowski, Kohlscheen, and A. Laqua.
Laboratoire d'Hematology, Hôpital Necker-Enfants Malades, Paris, France – V. Asnafi, L. Lhermitte, and E. Duroyon.
Department of Haematology/Oncology, Charles University, Prague, Czech Republic – J. Trka, O. Hrusak, and E. Mejstrikova.
Department of Pediatric Hematology and Oncology, Medical University of Silesia, Zabrze, Poland – T. Szczepanski, L. Sędek, and J. Bulsa.
Federal University of Rio de Janeiro, Rio de Janeiro, Brasil – E. Sobral da Costa and R. Mendonca de Pontes.
Dutch Childhood Oncology Group, Utrecht, The Netherlands – S. Nierkens, A. de Jong, and A. de Koning.
Department of Hematology, Cytometry Lab, Centro Hospitalar do Porto/University of Porto, Porto, Portugal – M. Lima and A. H. Santos.
Centro Ricerca Tettamanti, Clinica Pediatrica Università di Milano, Monza, Italy – G. Gaipa, C. Burracchi, and C. Bugarin.
Clinical Immunology Department, University Hospital La Paz-IdiPAZ, Madrid, Spain – E. Lopez-Granados and L. del Pino Molina.
Hematology Laboratory, CHU de Saint-Etienne, Saint-Etienne, France – L. Campos-Guyotat and C. Aanei.
Clinica Universidad de Navarra, Centro de Investigaciones Medicas Aplicadas, Universidad de Navarra, Pamplona, Spain – J. F. San Miguel and B. Paiva.
Unitat d’Hematopathologia, Hospital Clínic de Barcelona, Barcelona, Spain – N. Villamor-Casas and L. Magnano.
Laboratory of Clinical Biology, University Hospital Ghent, Ghent, Belgium – M. Hofmans, C. Bonroy, B. Denys, A. Willems, P. Breughe, and J. de Wolf.
Institute of Molecular Medicine, University of Lisbon, Lisbon, Portugal – A. E. Sousa and S. L. Silva.
Hemato-Oncology Unit, Centro Clinico Champalimaud, Champalimaud Foundation, Lisbon, Portugal – P. Lucio and C. João.

