

Key Points
Somatic mutations in chromatin- and cohesin-related genes, particularly STAG2, ASXL1, and DNMT3A, are common in GATA2 deficiency.
Mutations in other common myeloid malignancy–related genes, including splicing factors, are conspicuously rare in GATA2 deficiency.

Abstract
Patients with GATA2 deficiencyharbor de novo or inherited germline mutations in the GATA2 transcription factor gene, predisposing them to myeloid malignancies. There is considerable variation in disease progression, even among family members with the same mutation in GATA2. We investigated somatic mutations in 106 patients with GATA2 deficiency to identify acquired mutations that are associated with myeloid malignancies. Myelodysplastic syndrome (MDS) was the most common diagnosis (∼44%), followed by GATA2 bone marrow immunodeficiency disorder (G2BMID; ∼37%). Thirteen percent of the cohort had GATA2 mutations but displayed no disease manifestations. There were no correlations between age or sex with disease progression or survival. Cytogenetic analyses showed a high incidence of abnormalities (∼43%), notably trisomy 8 (∼23%) and monosomy 7 (∼12%), but the changes did not correlate with lower survival. Somatic mutations in ASXL1 and STAG2 were detected in ∼25% of patients, although the mutations were rarely concomitant. Mutations in DNMT3A were found in ∼10% of patients. These somatic mutations were found similarly in G2BMID and MDS, suggesting clonal hematopoiesis in early stages of disease, before the onset of MDS. ASXL1 mutations conferred a lower survival probability and were more prevalent in female patients. STAG2 mutations also conferred a lower survival probability, but did not show a statistically significant sex bias. There was a conspicuous absence of many commonly mutated genes associated with myeloid malignancies, including TET2, IDH1/2, and the splicing factor genes. Notably, somatic mutations in chromatin-related genes and cohesin genes characterized disease progression in GATA2 deficiency.
Introduction
GATA2 deficiency is a bone marrow failure syndrome resulting from germline mutations in the transcription factor gene GATA2. It was first described in 2010 as MonoMAC syndrome, to reflect 2 common disease manifestations: monocytopenia and Mycobacterium avium complex.1,2 It was also described as dendritic cell, monocyte, and B- and NK-cell lymphoid deficiency3 ; Emberger syndrome with lymphedema and monosomy 74 ; and familial myelodysplastic syndrome (MDS)/acute myelogenous leukemia (AML).5 Patients with GATA2 deficiency frequently present with a hypocellular bone marrow, monocytopenia and B- and NK-cell lymphopenia, and opportunistic infections, particularly human papilloma virus and mycobacterial infections.6,7 However, there is significant variability in other features of the disease, including peripheral lymphedema (Emberger syndrome), deafness, lupus-like autoimmunity, and pulmonary alveolar proteinosis, among others. The age at disease onset ranges considerably from pediatric to midlife, even within the same family, whereas other individuals with GATA2 mutations can remain asymptomatic throughout life. GATA2 deficiency penetrance is incomplete, with estimates ranging from ∼50% to 80%.8-10 Like penetrance, the rate of disease progression varies significantly, even among siblings in the same family, from a few years to decades.
Progression from immunodeficiency to hypocellular MDS is common in GATA2 deficiency, together with the appearance of cytogenetic abnormalities such as trisomy 8 and monosomy 7. Although a minority of patients progress to AML or proliferative chronic myelomonocytic leukemia (CMML), myeloid progression is an ominous development in GATA2 deficiency. In cases of non-GATA2 MDS and AML, disease progression is thought to be driven by the accumulation of somatic mutations. There is a consensus that a few dozen common mutations constitute a “hit list” for leukemia progression.11 These mutations tend to sort into 4 specific categories: transcription factors, chromatin modifiers, splicing factors, and signal transduction proteins. Somatic mutations in ASXL1, a chromatin modifier, are relatively common in non-GATA2 MDS/AML.11 Two reports on GATA2 deficiency noted a high frequency of ASXL1 mutations in patients with clonal progression.12,13 Recently, applications of genomic techniques in RUNX1, GATA2 deficiency, and CEBPA (CCAAT/enhancer-binding protein-alpha) mutations have suggested a high frequency of somatic ASXL1 mutations and have identified STAG2 as another commonly mutated gene, in contrast with a low frequency of other MDS/AML-associated genes.10
We applied next-generation sequencing techniques to blood and bone marrow samples of 106 patients with GATA2 deficiency, to identify acquired mutations that may drive myeloid progression of the disease. Genetic findings were compared with cytogenetics, biometric parameters, and GATA2 mutations. ASXL1, DNMT3A, and STAG2 mutations were the most common somatic ones; mutated BCOR and SETBP1 mutations were also recurrent, but less frequent. Mutations in the other most common genes associated with MDS/AML (TET2, IDH1/2 and the splicing factor genes) were notably rare or absent in GATA2 deficiency.
Methods
Patient samples
The clinical protocols were reviewed and approved by the Institutional Review Board of the National Institutes of Health, Clinical Research Center. The procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Declaration of Helsinki of 1975, as revised in 2008. All participants gave written informed consent. Patients in this study were enrolled in National Institute of Allergy and Infectious Diseases clinical trial 13-I-1057 (registered on clinicaltrials.gov as NCT01905826) and National Cancer Institute clinical trial 13C-0132 (NCT01861106). Peripheral blood and bone marrow samples were collected by using standard techniques. Clinical Laboratory Improvement Amendments–certified sequencing was performed on peripheral blood to identify GATA2 mutations. The GATA2 variant allele frequency (VAF) in coding mutations was >40% in DNA sequencing (described in the next section), consistent with germline mutations, either de novo or inherited (the exception was patient 100, for whom VAF data were not available). The patients were also evaluated for signs typical of MonoMAC syndrome to presume germline mutations. Genomic DNA (gDNA) for somatic mutation sequencing was isolated from peripheral blood leukocytes or bone marrow mononuclear cells by using the Puregene Blood Core Kit or DNeasy Blood and Tissue Kits (Qiagen Sciences, Germantown, MD). Standard metaphase cytogenetic analysis was performed on bone marrow aspirates. Survival probabilities were determined from Kaplan-Meier curves with significance determined with the Mantel-Cox log-rank test (Prism, version 9.0.0; GraphPad).
DNA sequencing
Sequencing was performed on gDNA isolated from peripheral blood leukocytes and/or bone marrow aspirates. Whole exome sequencing (WES) is described in the supplemental Methods. Targeted gene panel sequencing was performed with the Illumina TruSight Myeloid Panel, Integrated DNA Technologies xGen Lockdown Probes, or Qiagen Myeloid Neoplasms Panel (supplemental Table 1), and variants were called using the CCBR Pipeliner (Center for Cancer Research Collaborative Bioinformatics Resource; National Cancer Institute), omitting the deduplication filter. Details for sequencing, alignment, and quality control metrics are provided in the supplemental Methods. ASXL1 exons 12 and 13 (National Center of Biotechnology Information: NM_015338) were initially genotyped by direct DNA sequencing, as described previously.13 DNA sequences were analyzed with (MacVector, version 12.0; MacVector, Inc, Cary, NC). Mutations in ASXL1 were confirmed with independent polymerase chain reaction and primer sets. Statistical analyses and graphing were performed when indicated, using Prism, version 9.0.0. Statistical analyses were not performed when “n” was deemed too low.
Variant analysis
Somatic variants were analyzed by using MuTect2 with additional sorting parameters to exclude common population variants and variants of unknown significance. Variants were removed from further analysis based on a population VAF >0.1% in the ExAC/gnomAD or dbSNP (build 152) databases, sequencing read coverage of <20 reads by WES or <100 reads for targeted arrays, alternative read coverage of <5 reads by WES or <10 reads for targeted arrays, and/or <5% total reads. Mutations were considered to be pathogenic if damaging or deleterious scores by PolyPhen II or SIFT were consistent, or the Combined Annotation Dependent Depletion (CADD) score was >25, according to the CADD tool (https://cadd.gs.washington.edu).14 Variants were considered likely to be somatic if the VAF was <35% or was 70% on the X chromosome of the male participants. Other variants were considered probable germline and were not included in subsequent analyses. Variant bam files were also reviewed with the Integrative Genomics Viewer (IGV; version 2.6.2, http://software.broadinstitute.org), and additional sequence analysis was performed with MacVector, version 17.0.
Results
Patient population characteristics
We examined the biometrics and other parameters (age, sex, GATA2 mutation, diagnosis, cytogenetics, and survival) in 106 individuals bearing germline mutations in the GATA2 gene (n = 98) or with symptoms consistent with MonoMAC syndrome (n = 8; Table 1). These symptoms include hypocellular bone marrow with monocytopenia, B- and NK-cell lymphopenia and recurrent opportunistic infections. The total cohort consisted of 40 males (38%) and 66 females (62%) of similar age (32.7 ± 15.4 years) from 77 different families (Table 1; supplemental Figure 1). There were 42 different heterozygous GATA2 mutations from 72 different families and unidentified GATA2 mutations but with the MonoMAC syndrome from 5 families (Table 1). There were no patients with homozygous or compound heterozygous GATA2 mutations in this cohort.
Diagnoses, cytogenetics, and mutations in patients with GATA2 deficiency
| Pt | Family pedigree | GATA2 mutation | Disease | Cytogenetics | Sex | Inheritance | BMT | Survival | Common mutations | Total filtered variants |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 35.III.3 | c.1061C>T; p.T354M | MDS | Trisomy 8 | M | Familial | Yes | Deceased | 0 | 1 |
| 2 | 25.II.1 | c.1017 + 572C>T | G2BMID | Normal | M | Familial | No | Deceased | ASXL1, PTPN11, SMC1A, STAG2 | 5 |
| 3 | 8.I.1 | c.243_244delAinsGC; p.G82fs | G2BMID | Normal | M | De novo | Yes | Alive | DNMT3A, STAG2 | 4 |
| 4 | 12.I.1 | c.1084_1095del12; p.R362del4 | MDS | Monosomy 7, trisomy 21 | M | Unknown | Yes | Deceased | 0 | 10 |
| 5 | 17.I.1 | c.1061C>T; p.T354M | MDS | Trisomy 8 | M | Familial | Yes | Alive | 0 | 3 |
| 6 | 17.II.1 | c.1061C>T; p.T354M | G2BMID | Normal | M | Familial | Yes | Alive | STAG2 | 5 |
| 7 | 26.I.1 | c.302delG; p.G101fs | CMML | del(11)(q13q23) | F | Unknown | Yes | Deceased | ASXL1, CBL, DNMT3A | 13 |
| 8 | 34.II.2 | c.1116_1130del15; p.C373del5 | MDS | Monosomy 7 | M | Familial | Yes | Deceased | STAG2 | 5 |
| 9 | 34.I.1 | c.1116_1130del15; p.C373del5 | G2BMID | Normal | F | Familial | No | Alive | 0 | 5 |
| 10 | 31.II.2 | c.1187G>A; p.R396Q | G2BMID | Normal | M | Familial | Yes | Alive | STAG2 | 4 |
| 11 | 27.I.1 | c.586_593dup; p.G199fs | MDS | del(9)(q13q22) | F | Familial | Yes | Alive | ASXL1 | 6 |
| 12 | 39.I.2 | c.988C>T; p.R330X | MDS | Trisomy 8 | F | Familial | Yes | Alive | 0 | 4 |
| 13 | 39.I.1 | c.988C>T; p.R330X | MDS | Trisomy 8 | F | Familial | Yes | Alive | STAG2 | 2 |
| 14 | 15.I.1 | c.1186C>T; p.R396W | G2BMID | Normal | F | De novo | Yes | Alive | STAG2 | 4 |
| 15 | 38.I.1 | c.417dupT; p.V140Cfs | G2BMID | Normal | F | De novo | Yes | Alive | ASXL1, JAK2 | 12 |
| 16 | 9.II.1 | c.1192C>T; p.R398W | MDS | Normal | M | Familial | No | Deceased | BCOR, SRSF2, STAG* | 12 |
| 17 | 9.III.2 | c.1192C>T; p.R398W | Asymptomatic | Normal | M | Familial | No | Alive | 0 | 4 |
| 18 | 41.I.1 | c.1009C>T; p.R337X | G2BMID | Normal | F | De novo | Yes | Alive | ASXL1, DNMT3A, SMC1A | 14 |
| 19 | 42.I.1 | c.1186C>T; p.R396W | G2BMID | Normal | M | Familial | Yes | Alive | KDM2A | 2 |
| 20 | 14.I.1 | c.1187G>A; p.R396Q | G2BMID | Normal | F | De novo | No | Deceased | ASXL1, STAG2 | 9 |
| 21 | 146.I.1 | c.1082G>A; p.R361H | MDS | Normal | M | Familial | No | Deceased | DNMT3A, STAG2 | 7 |
| 22 | 159.I.1 | Unknown | AML | Trisomy 8, Monosomy 13 | F | Unknown | Yes | Deceased | KMT2A, MECOM | 14 |
| 23 | 48.II.11 | c.1017 + 572C>T | Asymptomatic | Normal | F | Familial | No | Alive | CUX1, DNMT3A, KMT2A,* U2 AF1 | 7 |
| 24 | 126.III.4† | Unknown | MDS | Trisomy 8 | F | Familial | Yes | Alive | 0 | 1 |
| 25 | 48.III.14 | c.1017 + 572C>T | G2BMID | der(1;14)(q10;p10), trisomy 21 | F | Familial | Yes | Alive | STAG2 | 1 |
| 26 | 126.III.23† | Unknown | MDS | Trisomy 8 | M | Familial | Yes | Alive | STAG2* | 3 |
| 27 | 126.II.8† | Unknown | Asymptomatic | Normal | F | Familial | No | Alive | 0 | 1 |
| 28 | 126.II.4† | Unknown | Asymptomatic | Normal | M | Familial | No | Alive | 0 | 0 |
| 29 | 256.I.1 | c.1123C>T; p.L375S | AML | Trisomy 8, trisomy 20 | F | De novo | Yes | Deceased | NRAS | 5 |
| 30 | 218.I.1 | c.1114G>A; p.A372T | MDS | Trisomy 8 | F | Fe novo | No | Deceased | DNMT3A | 10 |
| 31 | 129.I.1 | c.802G>T; p.G268X | MDS | del(13)(q12q14) | F | Unknown | No | Alive | 0 | 2 |
| 32 | 233.I.1 | c.803delG; p.G268fs | G2BMID | Normal | F | De novo | Yes | Alive | 0 | 11 |
| 33 | 203.I.1 | c.1021delG; p.A341Pfs | G2BMID | Normal | M | Familial | Yes | Alive | DNMT3A | 6 |
| 34 | 47.I.1 | c.1084C>T; p.R362X | Asymptomatic | Normal | F | Familial | No | Alive | 0 | 1 |
| 35 | 270.I.1 | c.1082G>A; p.R361H | G2BMID | Normal | M | Familial | Yes | Alive | BCOR | 7 |
| 36 | 49.III.2 | c.1084C>T; p.R362X | G2BMID | Normal | F | Familial | Yes | Alive | 0 | 3 |
| 37 | 281.I.1 | c.1186C>T; p.R396W | MDS | inv(9)(p12q13) | F | Unknown | Yes | Deceased | ASXL1 | 5 |
| 38 | 50.II.2 | c.1128C>A; p.Y376X | MDS | Normal | F | Familial | Yes | Alive | STAG2 | 3 |
| 39 | 50.II.1 | c.1128C>A; p.Y376X | MDS | der(1;7)(q10;p10), trisomy 8 | M | Familial | Yes | Alive | STAG2 | 14 |
| 40 | 293.I.1 | Unknown | AML | der(5) t(5;13)(q13q13) | M | Unknown | Yes | Alive | 0 | 5 |
| 41 | 291.I.1 | c.1084C>T; p.R362X | MDS | Monosomy 7 | M | De novo | Yes | Alive | STAG2 | 2 |
| 42 | 216.I.1 | c.1192C>T; p.R398W | MDS | der(1;7)(q10;p10), 1+der(1;13)(q10;q10), trisomy 8, monosomy X | F | Unknown | Yes | Alive | ASXL1 | 7 |
| 43 | 51.III.1 | c.1192C>T; p.R398W | G2BMID | Normal | F | Familial | Yes | Deceased | ASXL1 | 6 |
| 44 | 51.II.1 | c.1192C>T; p.R398W | Asymptomatic | Normal | M | Familial | No | Alive | 0 | 2 |
| 45 | 52.II.4 | c.1082G>C; p.R361P | G2BMID | Normal | F | Familial | No | Alive | BCL9 | 2 |
| 46 | 52.II.5 | c.1082G>C; p.R361P | G2BMID | Normal | F | Familial | No | Alive | 0 | 4 |
| 47 | 52.I.4 | c.1082G>C; p.R361P | MDS | Normal | M | Familial | Yes | Deceased | STAG2 | 4 |
| 48 | 330.I.1 | c.898dupG; p.A300Gfs | MDS | Normal | F | Familial | Yes | Deceased | ASXL1, BCOR | 5 |
| 49 | 335.III.1 | c.1017 + 572C>T | MDS | Normal | F | Familial | Yes | Alive | 0 | 1 |
| 50 | 340.II.1 | c.1159_1160dupAC; p.M388fs | G2BMID | Monosomy 7, der(1;7)(q10;p10), del(13)(q12q22) | M | Familial | Yes | Alive | STAG2‡ | 3 |
| 51 | 324.I.1 | c.1018del7; p.S340fs | MDS | Trisomy 8 | F | De novo | Yes | Alive | STAG2 | 6 |
| 52 | 342.I.1 | c.1017 + 572C>T | MDS | Monosomy 7 | M | Unknown | Yes | Alive | SAMD9, SETBP1, U2AF1 | 7 |
| 53 | 53.V.4 | c.1017 + 572C>T | G2BMID | Normal | F | Familial | No | Alive | 0 | 2 |
| 54 | 53.V.3 | c.1017 + 572C>T | MDS | Normal | F | Familial | Yes | Alive | ETV6 | 3 |
| 55 | 350.I.1 | c.248delA; p.Q83Rfs‡ | MDS | Normal | F | De novo | Yes | Alive | 0 | 4 |
| 56 | 349.II.4 | c.1192C>T; p.R398W | MDS | trp(1)(q21q32) | F | Familial | No | Alive | DNMT3A,* U2 AF1 | 11 |
| 57 | 347.I.1 | c.1017 + 2T>A | MDS | Normal | F | De novo | Yes | Alive | STAG2 | 5 |
| 58 | 351.I.1 | c.1192C>T; p.R398W | MDS | Normal | F | De novo | Yes | Alive | 0 | 2 |
| 59 | 333.II.3 | c.1018-50_1143 + 247del; p.S340-N381del | MDS | Trisomy 8 | M | Familial | Yes | Alive | DNMT3A, STAG2* | 6 |
| 60 | 337.I.1 | c.921dupG; p.R308Afs‡ | G2BMID | Normal | F | Unknown | Yes | Alive | STAG2 | 12 |
| 61 | 360.I.1 | c.1024_1025insGCCG; p.A342Gfs | G2BMID | Normal | F | Unknown | Yes | Alive | STAG2 | 2 |
| 62 | 357.II.1 | c.1017 + 1G>T‡ | MDS | Monosomy 7 | F | Familial | Yes | Alive | RUNX1 | 4 |
| 63 | 357.I.1 | c.1017 + 1G>T‡ | MDS | Trisomy 8 | F | Familial | No | Alive | ASXL1 | 6 |
| 64 | 362.I.1 | c.247C>T; p.Q83X‡ | MDS | Monosomy 7 | F | De novo | Yes | Alive | STAG2* | 5 |
| 65 | n/a | c.1084C>T; p.R362X | G2BMID | Normal | F | De novo | No | Deceased | NOTCH2 | 6 |
| 66 | n/a | c.680_683del; p.S227fs‡ | G2BMID | Normal | F | De novo | Yes | Alive | STAG2 | 4 |
| 67 | 365.I.1 | c.1081C>T; p.R361C | MDS | der(1;7)(q10;p10), trisomy 8 | M | Familial | Yes | Alive | STAG2* | 7 |
| 68 | 368.I.1 | c.1061C>T; p.T354M | Asymptomatic | Unknown | F | Familial | No | Alive | DNMT3A | 7 |
| 69 | 370.I.1 | unknown | MDS | Normal | F | Unknown | Yes | Deceased | ASXL1, RUNX1 | 10 |
| 70 | 375.I.2 | c.1192C>T; p.R398W | MDS | (1)t(1;15), Trisomy 8 | F | Familial | Yes | Alive | ASXL1, CUX1, RAD21 | 10 |
| 71 | n/a | c.1150delA; p.R384fs‡ | G2BMID | Normal | M | De novo | Yes | Alive | 0 | 2 |
| 72 | 378.I.1 | c.1021delG; p.A341Pfs | MDS | Normal | M | unknown | Yes | Alive | BCOR | 5 |
| 73 | 379.I.1 | c.1277C>G; p.S426C‡ | G2BMID | Normal | F | Unknown | No | Alive | 0 | 3 |
| 74 | 367.I.1 | unknown | MDS | Trisomy 8 | F | Unknown | Yes | Alive | 0 | 2 |
| 75 | 382.I.2 | c.1061C>A; p.T354K | MDS | Normal | F | Familial | Yes | Alive | 0 | 7 |
| 76 | 283.II.1 | c.58C>T; p.Q20X‡ | MDS | der(1;7)(q10:p10) | F | Familial | Yes | Alive | SETBP1, STAG2 | 5 |
| 77 | n/a | c.1061C>T; p.T354M | G2BMID | Normal | F | De novo | Yes | Alive | 0 | 3 |
| 78 | 4.III.5 | c.1017 + 572C>T | CMML | Monosomy 7 | M | familial | Yes | Alive | ASXL1, SETBP1, U2AF1 | 26 |
| 79 | 384.I.1 | c.1192C>T; p.R398W | MDS | Monosomy 7 | F | Familial | Yes | Alive | ASXL1 | 4 |
| 80 | n/a | c.1192C>T; p.R398W | G2BMID | Normal | F | Familial | No | Alive | 0 | 2 |
| 81 | 393.II.1 | c.1061C>T; p.T354M | G2BMID | Normal | F | Familial | No | Alive | 0 | 9 |
| 82 | n/a | c.840delT; p.K281Sfs‡ | G2BMID | Normal | F | De novo | Yes | Alive | 0 | 3 |
| 83 | 390.I.1 | c.1009C>T; p.R337X | G2BMID | Normal | F | De novo | Yes | Alive | ASXL1, STAG2 | 4 |
| 84 | 394.I.1 | c.1114G>A; p.A372T | G2BMID | Normal | M | Unknown | Yes | Alive | 0 | 6 |
| 85 | 349.IV.5 | c.1192C>T; p.R398W | G2BMID | Normal | M | Familial | Yes | Alive | 0 | 1 |
| 86 | 349.III.16 | c.1192C>T; p.R398W | MDS | Trisomy 8 | F | Familial | Yes | Alive | 0 | 4 |
| 87 | 349.IV.7 | c.1192C>T; p.R398W | G2BMID | Normal | F | Familial | No | Alive | 0 | 4 |
| 88 | 1.IV.1 | c.1192C>T; p.R398W | G2BMID | Normal | F | Familial | Yes | Alive | 0 | 3 |
| 89 | 1.III.2 | c.1192C>T; p.R398W | Asymptomatic | Normal | M | Familial | No | Alive | DNMT3A | 5 |
| 90 | 389.I.1 | c.1024_1025insG; p.A342Gfs | G2BMID | Normal | F | Unknown | Yes | Alive | 0 | 8 |
| 91 | 4.III.2 | c.1017 + 572C>T | G2BMID | Normal | F | Familial | No | Alive | 0 | NA |
| 92 | 37.I.1 | c.1081C>T, p.R361C | MDS | Normal | F | De novo | Yes | Alive | DNMT3A, FLT3 | NA |
| 93 | 48.II.6 | c.1017 + 572C>T | Asymptomatic | Normal | F | Familial | No | Alive | 0 | NA |
| 94 | 48.II.2 | c.1017 + 572C>T | Asymptomatic | Normal | F | Familial | No | Alive | 0 | NA |
| 95 | n/a | c.1187G>A; p.R396Q | MDS | Normal | F | Familial | Yes | Alive | ASXL1 | NA |
| 96 | 4.III.1 | c.1017 + 572C>T | Asymptomatic | Normal | M | Familial | No | Alive | 0 | NA |
| 97 | 4.I.1 | c.1017 + 572C>T | CMML | Normal | M | Familial | No | Alive | 0 | NA |
| 98 | 17.II.2 | c.1061C>T; p.T354M | G2BMID | Normal | M | Familial | No | Alive | 0 | NA |
| 99 | 48.II.8 | c.1017 + 572C>T | Asymptomatic | Normal | M | Familial | No | Alive | 0 | NA |
| 100 | 4.III.4 | c.1017 + 572C>T | MDS | Monosomy 7, Trisomy 8 | M | Familial | Yes | Alive | 0 | NA |
| 101 | n/a | c.839delC; p.P280Lfs | MDS | Trisomy 8 | M | Unknown | No | Alive | 0 | NA |
| 102 | 6.I.1 | c.1017 + 512del28 | Asymptomatic | Normal | M | Familial | No | Alive | 0 | NA |
| 103 | 335.II.1 | c.1017 + 572C>T | Asymptomatic | Normal | F | Familial | No | Alive | 0 | NA |
| 104 | n/a | c.1084C>T; p.R362X | G2BMID | Normal | M | De novo | No | Alive | STAG2* | 5 |
| 105 | 277.I.1 | c.1187G>A; p.R396Q | MDS | Trisomy 8 | F | De novo | Yes | Alive | 0 | 4 |
| 106 | 368.II.1 | c.1061C>T; p.T354M | MDS | Monosomy 7 | M | Familial | Yes | Alive | 0 | NA |
| Pt | Family pedigree | GATA2 mutation | Disease | Cytogenetics | Sex | Inheritance | BMT | Survival | Common mutations | Total filtered variants |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 35.III.3 | c.1061C>T; p.T354M | MDS | Trisomy 8 | M | Familial | Yes | Deceased | 0 | 1 |
| 2 | 25.II.1 | c.1017 + 572C>T | G2BMID | Normal | M | Familial | No | Deceased | ASXL1, PTPN11, SMC1A, STAG2 | 5 |
| 3 | 8.I.1 | c.243_244delAinsGC; p.G82fs | G2BMID | Normal | M | De novo | Yes | Alive | DNMT3A, STAG2 | 4 |
| 4 | 12.I.1 | c.1084_1095del12; p.R362del4 | MDS | Monosomy 7, trisomy 21 | M | Unknown | Yes | Deceased | 0 | 10 |
| 5 | 17.I.1 | c.1061C>T; p.T354M | MDS | Trisomy 8 | M | Familial | Yes | Alive | 0 | 3 |
| 6 | 17.II.1 | c.1061C>T; p.T354M | G2BMID | Normal | M | Familial | Yes | Alive | STAG2 | 5 |
| 7 | 26.I.1 | c.302delG; p.G101fs | CMML | del(11)(q13q23) | F | Unknown | Yes | Deceased | ASXL1, CBL, DNMT3A | 13 |
| 8 | 34.II.2 | c.1116_1130del15; p.C373del5 | MDS | Monosomy 7 | M | Familial | Yes | Deceased | STAG2 | 5 |
| 9 | 34.I.1 | c.1116_1130del15; p.C373del5 | G2BMID | Normal | F | Familial | No | Alive | 0 | 5 |
| 10 | 31.II.2 | c.1187G>A; p.R396Q | G2BMID | Normal | M | Familial | Yes | Alive | STAG2 | 4 |
| 11 | 27.I.1 | c.586_593dup; p.G199fs | MDS | del(9)(q13q22) | F | Familial | Yes | Alive | ASXL1 | 6 |
| 12 | 39.I.2 | c.988C>T; p.R330X | MDS | Trisomy 8 | F | Familial | Yes | Alive | 0 | 4 |
| 13 | 39.I.1 | c.988C>T; p.R330X | MDS | Trisomy 8 | F | Familial | Yes | Alive | STAG2 | 2 |
| 14 | 15.I.1 | c.1186C>T; p.R396W | G2BMID | Normal | F | De novo | Yes | Alive | STAG2 | 4 |
| 15 | 38.I.1 | c.417dupT; p.V140Cfs | G2BMID | Normal | F | De novo | Yes | Alive | ASXL1, JAK2 | 12 |
| 16 | 9.II.1 | c.1192C>T; p.R398W | MDS | Normal | M | Familial | No | Deceased | BCOR, SRSF2, STAG* | 12 |
| 17 | 9.III.2 | c.1192C>T; p.R398W | Asymptomatic | Normal | M | Familial | No | Alive | 0 | 4 |
| 18 | 41.I.1 | c.1009C>T; p.R337X | G2BMID | Normal | F | De novo | Yes | Alive | ASXL1, DNMT3A, SMC1A | 14 |
| 19 | 42.I.1 | c.1186C>T; p.R396W | G2BMID | Normal | M | Familial | Yes | Alive | KDM2A | 2 |
| 20 | 14.I.1 | c.1187G>A; p.R396Q | G2BMID | Normal | F | De novo | No | Deceased | ASXL1, STAG2 | 9 |
| 21 | 146.I.1 | c.1082G>A; p.R361H | MDS | Normal | M | Familial | No | Deceased | DNMT3A, STAG2 | 7 |
| 22 | 159.I.1 | Unknown | AML | Trisomy 8, Monosomy 13 | F | Unknown | Yes | Deceased | KMT2A, MECOM | 14 |
| 23 | 48.II.11 | c.1017 + 572C>T | Asymptomatic | Normal | F | Familial | No | Alive | CUX1, DNMT3A, KMT2A,* U2 AF1 | 7 |
| 24 | 126.III.4† | Unknown | MDS | Trisomy 8 | F | Familial | Yes | Alive | 0 | 1 |
| 25 | 48.III.14 | c.1017 + 572C>T | G2BMID | der(1;14)(q10;p10), trisomy 21 | F | Familial | Yes | Alive | STAG2 | 1 |
| 26 | 126.III.23† | Unknown | MDS | Trisomy 8 | M | Familial | Yes | Alive | STAG2* | 3 |
| 27 | 126.II.8† | Unknown | Asymptomatic | Normal | F | Familial | No | Alive | 0 | 1 |
| 28 | 126.II.4† | Unknown | Asymptomatic | Normal | M | Familial | No | Alive | 0 | 0 |
| 29 | 256.I.1 | c.1123C>T; p.L375S | AML | Trisomy 8, trisomy 20 | F | De novo | Yes | Deceased | NRAS | 5 |
| 30 | 218.I.1 | c.1114G>A; p.A372T | MDS | Trisomy 8 | F | Fe novo | No | Deceased | DNMT3A | 10 |
| 31 | 129.I.1 | c.802G>T; p.G268X | MDS | del(13)(q12q14) | F | Unknown | No | Alive | 0 | 2 |
| 32 | 233.I.1 | c.803delG; p.G268fs | G2BMID | Normal | F | De novo | Yes | Alive | 0 | 11 |
| 33 | 203.I.1 | c.1021delG; p.A341Pfs | G2BMID | Normal | M | Familial | Yes | Alive | DNMT3A | 6 |
| 34 | 47.I.1 | c.1084C>T; p.R362X | Asymptomatic | Normal | F | Familial | No | Alive | 0 | 1 |
| 35 | 270.I.1 | c.1082G>A; p.R361H | G2BMID | Normal | M | Familial | Yes | Alive | BCOR | 7 |
| 36 | 49.III.2 | c.1084C>T; p.R362X | G2BMID | Normal | F | Familial | Yes | Alive | 0 | 3 |
| 37 | 281.I.1 | c.1186C>T; p.R396W | MDS | inv(9)(p12q13) | F | Unknown | Yes | Deceased | ASXL1 | 5 |
| 38 | 50.II.2 | c.1128C>A; p.Y376X | MDS | Normal | F | Familial | Yes | Alive | STAG2 | 3 |
| 39 | 50.II.1 | c.1128C>A; p.Y376X | MDS | der(1;7)(q10;p10), trisomy 8 | M | Familial | Yes | Alive | STAG2 | 14 |
| 40 | 293.I.1 | Unknown | AML | der(5) t(5;13)(q13q13) | M | Unknown | Yes | Alive | 0 | 5 |
| 41 | 291.I.1 | c.1084C>T; p.R362X | MDS | Monosomy 7 | M | De novo | Yes | Alive | STAG2 | 2 |
| 42 | 216.I.1 | c.1192C>T; p.R398W | MDS | der(1;7)(q10;p10), 1+der(1;13)(q10;q10), trisomy 8, monosomy X | F | Unknown | Yes | Alive | ASXL1 | 7 |
| 43 | 51.III.1 | c.1192C>T; p.R398W | G2BMID | Normal | F | Familial | Yes | Deceased | ASXL1 | 6 |
| 44 | 51.II.1 | c.1192C>T; p.R398W | Asymptomatic | Normal | M | Familial | No | Alive | 0 | 2 |
| 45 | 52.II.4 | c.1082G>C; p.R361P | G2BMID | Normal | F | Familial | No | Alive | BCL9 | 2 |
| 46 | 52.II.5 | c.1082G>C; p.R361P | G2BMID | Normal | F | Familial | No | Alive | 0 | 4 |
| 47 | 52.I.4 | c.1082G>C; p.R361P | MDS | Normal | M | Familial | Yes | Deceased | STAG2 | 4 |
| 48 | 330.I.1 | c.898dupG; p.A300Gfs | MDS | Normal | F | Familial | Yes | Deceased | ASXL1, BCOR | 5 |
| 49 | 335.III.1 | c.1017 + 572C>T | MDS | Normal | F | Familial | Yes | Alive | 0 | 1 |
| 50 | 340.II.1 | c.1159_1160dupAC; p.M388fs | G2BMID | Monosomy 7, der(1;7)(q10;p10), del(13)(q12q22) | M | Familial | Yes | Alive | STAG2‡ | 3 |
| 51 | 324.I.1 | c.1018del7; p.S340fs | MDS | Trisomy 8 | F | De novo | Yes | Alive | STAG2 | 6 |
| 52 | 342.I.1 | c.1017 + 572C>T | MDS | Monosomy 7 | M | Unknown | Yes | Alive | SAMD9, SETBP1, U2AF1 | 7 |
| 53 | 53.V.4 | c.1017 + 572C>T | G2BMID | Normal | F | Familial | No | Alive | 0 | 2 |
| 54 | 53.V.3 | c.1017 + 572C>T | MDS | Normal | F | Familial | Yes | Alive | ETV6 | 3 |
| 55 | 350.I.1 | c.248delA; p.Q83Rfs‡ | MDS | Normal | F | De novo | Yes | Alive | 0 | 4 |
| 56 | 349.II.4 | c.1192C>T; p.R398W | MDS | trp(1)(q21q32) | F | Familial | No | Alive | DNMT3A,* U2 AF1 | 11 |
| 57 | 347.I.1 | c.1017 + 2T>A | MDS | Normal | F | De novo | Yes | Alive | STAG2 | 5 |
| 58 | 351.I.1 | c.1192C>T; p.R398W | MDS | Normal | F | De novo | Yes | Alive | 0 | 2 |
| 59 | 333.II.3 | c.1018-50_1143 + 247del; p.S340-N381del | MDS | Trisomy 8 | M | Familial | Yes | Alive | DNMT3A, STAG2* | 6 |
| 60 | 337.I.1 | c.921dupG; p.R308Afs‡ | G2BMID | Normal | F | Unknown | Yes | Alive | STAG2 | 12 |
| 61 | 360.I.1 | c.1024_1025insGCCG; p.A342Gfs | G2BMID | Normal | F | Unknown | Yes | Alive | STAG2 | 2 |
| 62 | 357.II.1 | c.1017 + 1G>T‡ | MDS | Monosomy 7 | F | Familial | Yes | Alive | RUNX1 | 4 |
| 63 | 357.I.1 | c.1017 + 1G>T‡ | MDS | Trisomy 8 | F | Familial | No | Alive | ASXL1 | 6 |
| 64 | 362.I.1 | c.247C>T; p.Q83X‡ | MDS | Monosomy 7 | F | De novo | Yes | Alive | STAG2* | 5 |
| 65 | n/a | c.1084C>T; p.R362X | G2BMID | Normal | F | De novo | No | Deceased | NOTCH2 | 6 |
| 66 | n/a | c.680_683del; p.S227fs‡ | G2BMID | Normal | F | De novo | Yes | Alive | STAG2 | 4 |
| 67 | 365.I.1 | c.1081C>T; p.R361C | MDS | der(1;7)(q10;p10), trisomy 8 | M | Familial | Yes | Alive | STAG2* | 7 |
| 68 | 368.I.1 | c.1061C>T; p.T354M | Asymptomatic | Unknown | F | Familial | No | Alive | DNMT3A | 7 |
| 69 | 370.I.1 | unknown | MDS | Normal | F | Unknown | Yes | Deceased | ASXL1, RUNX1 | 10 |
| 70 | 375.I.2 | c.1192C>T; p.R398W | MDS | (1)t(1;15), Trisomy 8 | F | Familial | Yes | Alive | ASXL1, CUX1, RAD21 | 10 |
| 71 | n/a | c.1150delA; p.R384fs‡ | G2BMID | Normal | M | De novo | Yes | Alive | 0 | 2 |
| 72 | 378.I.1 | c.1021delG; p.A341Pfs | MDS | Normal | M | unknown | Yes | Alive | BCOR | 5 |
| 73 | 379.I.1 | c.1277C>G; p.S426C‡ | G2BMID | Normal | F | Unknown | No | Alive | 0 | 3 |
| 74 | 367.I.1 | unknown | MDS | Trisomy 8 | F | Unknown | Yes | Alive | 0 | 2 |
| 75 | 382.I.2 | c.1061C>A; p.T354K | MDS | Normal | F | Familial | Yes | Alive | 0 | 7 |
| 76 | 283.II.1 | c.58C>T; p.Q20X‡ | MDS | der(1;7)(q10:p10) | F | Familial | Yes | Alive | SETBP1, STAG2 | 5 |
| 77 | n/a | c.1061C>T; p.T354M | G2BMID | Normal | F | De novo | Yes | Alive | 0 | 3 |
| 78 | 4.III.5 | c.1017 + 572C>T | CMML | Monosomy 7 | M | familial | Yes | Alive | ASXL1, SETBP1, U2AF1 | 26 |
| 79 | 384.I.1 | c.1192C>T; p.R398W | MDS | Monosomy 7 | F | Familial | Yes | Alive | ASXL1 | 4 |
| 80 | n/a | c.1192C>T; p.R398W | G2BMID | Normal | F | Familial | No | Alive | 0 | 2 |
| 81 | 393.II.1 | c.1061C>T; p.T354M | G2BMID | Normal | F | Familial | No | Alive | 0 | 9 |
| 82 | n/a | c.840delT; p.K281Sfs‡ | G2BMID | Normal | F | De novo | Yes | Alive | 0 | 3 |
| 83 | 390.I.1 | c.1009C>T; p.R337X | G2BMID | Normal | F | De novo | Yes | Alive | ASXL1, STAG2 | 4 |
| 84 | 394.I.1 | c.1114G>A; p.A372T | G2BMID | Normal | M | Unknown | Yes | Alive | 0 | 6 |
| 85 | 349.IV.5 | c.1192C>T; p.R398W | G2BMID | Normal | M | Familial | Yes | Alive | 0 | 1 |
| 86 | 349.III.16 | c.1192C>T; p.R398W | MDS | Trisomy 8 | F | Familial | Yes | Alive | 0 | 4 |
| 87 | 349.IV.7 | c.1192C>T; p.R398W | G2BMID | Normal | F | Familial | No | Alive | 0 | 4 |
| 88 | 1.IV.1 | c.1192C>T; p.R398W | G2BMID | Normal | F | Familial | Yes | Alive | 0 | 3 |
| 89 | 1.III.2 | c.1192C>T; p.R398W | Asymptomatic | Normal | M | Familial | No | Alive | DNMT3A | 5 |
| 90 | 389.I.1 | c.1024_1025insG; p.A342Gfs | G2BMID | Normal | F | Unknown | Yes | Alive | 0 | 8 |
| 91 | 4.III.2 | c.1017 + 572C>T | G2BMID | Normal | F | Familial | No | Alive | 0 | NA |
| 92 | 37.I.1 | c.1081C>T, p.R361C | MDS | Normal | F | De novo | Yes | Alive | DNMT3A, FLT3 | NA |
| 93 | 48.II.6 | c.1017 + 572C>T | Asymptomatic | Normal | F | Familial | No | Alive | 0 | NA |
| 94 | 48.II.2 | c.1017 + 572C>T | Asymptomatic | Normal | F | Familial | No | Alive | 0 | NA |
| 95 | n/a | c.1187G>A; p.R396Q | MDS | Normal | F | Familial | Yes | Alive | ASXL1 | NA |
| 96 | 4.III.1 | c.1017 + 572C>T | Asymptomatic | Normal | M | Familial | No | Alive | 0 | NA |
| 97 | 4.I.1 | c.1017 + 572C>T | CMML | Normal | M | Familial | No | Alive | 0 | NA |
| 98 | 17.II.2 | c.1061C>T; p.T354M | G2BMID | Normal | M | Familial | No | Alive | 0 | NA |
| 99 | 48.II.8 | c.1017 + 572C>T | Asymptomatic | Normal | M | Familial | No | Alive | 0 | NA |
| 100 | 4.III.4 | c.1017 + 572C>T | MDS | Monosomy 7, Trisomy 8 | M | Familial | Yes | Alive | 0 | NA |
| 101 | n/a | c.839delC; p.P280Lfs | MDS | Trisomy 8 | M | Unknown | No | Alive | 0 | NA |
| 102 | 6.I.1 | c.1017 + 512del28 | Asymptomatic | Normal | M | Familial | No | Alive | 0 | NA |
| 103 | 335.II.1 | c.1017 + 572C>T | Asymptomatic | Normal | F | Familial | No | Alive | 0 | NA |
| 104 | n/a | c.1084C>T; p.R362X | G2BMID | Normal | M | De novo | No | Alive | STAG2* | 5 |
| 105 | 277.I.1 | c.1187G>A; p.R396Q | MDS | Trisomy 8 | F | De novo | Yes | Alive | 0 | 4 |
| 106 | 368.II.1 | c.1061C>T; p.T354M | MDS | Monosomy 7 | M | Familial | Yes | Alive | 0 | NA |
Patient (Pt) number and Family pedigree designation are listed chronologically as the samples were collected. The survival time is from first sample collection until the end of the study. Common mutations are those found in ≥3 patients in this study or >5% in other studies of myeloid malignancies. Mutations in patient 78 have also been described in Molina et al.67 NA indicates that WES was not performed (supplemental Table 1).
BMT, bone marrow transplant.
Two different variants were found in the gene for the indicated patient.
The pedigree for family 126 is given in supplemental Figure 3.
GATA2 mutations unique to our study.
The patients were divided into 5 separate categories based on their bone marrow and clinical status: asymptomatic (Asym), GATA2 bone marrow immunodeficiency disorder (G2BMID),6 MDS, AML, and CMML (Figure 1A). Asymptomatic individuals had normal complete blood counts (CBCs), normal bone marrow pathology, and no history of recurrent or life-threatening infections. Patients with G2BMID had a hypocellular marrow with cytopenia, but no definitive evidence of dysplasia. MDS was characterized by dysplasia, ineffective hematopoiesis, and other associated factors, including abnormal cytogenetics. Patients with AML or CMML were frequently grouped together and are designated AML/CMML for analysis because of low sample numbers.
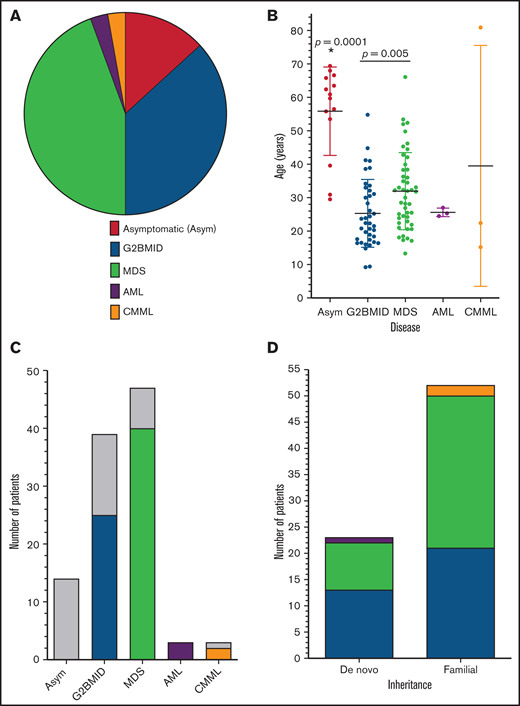
Patients with GATA2 deficiency by diagnosis, age and inheritance. (A) Bone marrow diagnosis. The relative frequency of bone marrow diagnoses among patients with GATA2 deficiency/MonoMAC (n = 106). (B) Diagnosis vs age. Plot of patient age at sample collection for each diagnosis category. The means and standard deviations are shown: Asym n = 14, G2BMID n = 39, MDS n = 47, AML n = 3, CMML n = 3. *Significant differences by ANOVA; P-values, by Student t test. (C) Patients with HSCT for each diagnosis category. Patients with HSCT are indicated according to diagnosis and color in the legend to panel A and those without are in gray. (D) Diagnosis vs mode of inheritance. The number of patients with de novo or familial inheritance with each bone marrow diagnosis as indicated by the color scheme.
Patients with GATA2 deficiency by diagnosis, age and inheritance. (A) Bone marrow diagnosis. The relative frequency of bone marrow diagnoses among patients with GATA2 deficiency/MonoMAC (n = 106). (B) Diagnosis vs age. Plot of patient age at sample collection for each diagnosis category. The means and standard deviations are shown: Asym n = 14, G2BMID n = 39, MDS n = 47, AML n = 3, CMML n = 3. *Significant differences by ANOVA; P-values, by Student t test. (C) Patients with HSCT for each diagnosis category. Patients with HSCT are indicated according to diagnosis and color in the legend to panel A and those without are in gray. (D) Diagnosis vs mode of inheritance. The number of patients with de novo or familial inheritance with each bone marrow diagnosis as indicated by the color scheme.
MDS was the most common bone marrow diagnosis in this cohort (44.3%; n = 47), followed by G2BMID (36.8%; n = 39), normal (13.2%; n = 14), and AML/CMML (5.7%; n = 6; Figure 1A). Age ranged from 9.4 to 80.9 years, and clinical status ranged from asymptomatic to advanced proliferative leukemia (Figure 1B). Patients with MDS were significantly older (32.0 ± 11.4 years) than those with G2BMID (25.3 ± 10.2 years; Figure 1B). This study also included 6 patients with overt leukemia: 3 with AML and 3 with proliferative CMML. Together, the patients with AML and CMML were younger than those with MDS, with the exception of an 81-year-old man with CMML. The diagnoses were not significantly biased by sex (supplemental Figure 1).
Fourteen patients had pathogenic GATA2 mutations or were carriers of MonoMAC syndrome based on an extended family pedigree (supplemental Figure 2), but these patients remained asymptomatic throughout the course of the study. The asymptomatic group was significantly older than the group with clinical symptoms, with an average age of 55.9 ± 13.2 years vs 29.2 ± 12.3 years (n = 92; Figure 1B).
Bone marrow transplant
Hematopoietic stem cell transplant (HSCT) represents the definitive treatment of GATA2 deficiency. Seventy-six percent of the patients (70 of 92) underwent HSCT (Figure 1C), and 84% (59 of 70) were alive and disease-free after HSCT. Of the 24% (22 of 92) of affected patients who did not receive transplants, 77% (17 of 22) were alive at the end of the study (Table 1). Detailed descriptions of HSCT in patients with GATA2 deficiency are presented elsewhere (Parta et al15 and “HSCT with GATA2 deficiency: influence of donor stem cell source and posttransplantation cyclophosphamide”; Nichols-Vinueza et al.16 ).
Inheritance
GATA2 deficiency may be inherited through an existing germline mutation in a parent or as a de novo germline mutation first expressed in the patient (see “Methods”). The GATA2 mutation inheritance pattern was investigated as a factor for disease development and progression. The patients in this cohort were classified as familial if they had a first-degree relative with the same GATA2 mutation or de novo if neither biological parent tested positive for a patient’s GATA2 mutation. Cases that did not have the data for either of these scenarios were classified as “unknown.” An inheritance pattern was established for 89 patients in 60 families. Familial inheritance was observed in 62% (37 of 60) and de novo inheritance in 38% (23 of 60; Figure 1D) of the cases. The inheritance mode could not be confirmed in 17 families. The inheritance mechanism did not affect the bone marrow diagnosis (Table 1; Figures 1D and 4A).
Cytogenetics
Cytogenetic analysis remains a key diagnostic component in myeloid malignancies. Cytogenetic analysis was performed on samples from 92 patients with disease manifestations, and at least 1 abnormality was found in 43% of the cases (Table 1). The most common abnormalities were trisomy 8 (23%; 21 of 92) and monosomy 7 (12%; 11 of 92; Figure 2A). Other cytogenetic abnormalities were found in 18% (16 of 92) of patients, including 5 with der(1;7)(q10;p10). The other cytogenetic abnormalities were concurrent with trisomy 8 in 6 cases and monosomy 7 in 1 case. The presence of more than 1 type of cytogenetic abnormality was found in 11% of patients (10 of 92).
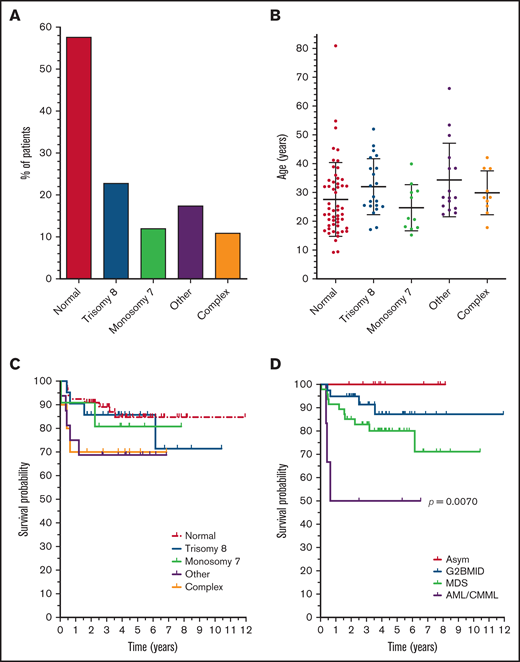
Cytogenetic abnormalities in GATA2 deficiency. (A) Cytogenetic findings. Percentages of patients with the indicated cytogenetic abnormalities. The number of patients with trisomy 8 or monosomy 7 includes all patients with this abnormality, independent of other abnormalities. “Other,” abnormalities other than trisomy 8 or monosomy 7; “complex,” more than 1 abnormality in the same patient, including trisomy 8 and/or monosomy 7. (B) Cytogenetics vs age. The age distribution of cytogenetic findings, with the mean ages and standard deviations indicated (ANOVA, P = .1069). (C) Cytogenetics vs survival. Kaplan-Meier survival probability as a function of cytogenetics. Classifications are as in panel A. Survival is from the initial sample collection to the end of the study. (D) Diagnosis vs survival. Kaplan-Meier survival probability as a function of bone marrow diagnosis. Patients with AML or CMML were combined because of the low number of cases. Each graph shows the survival time from sample collection until the end point. Mantel-Cox test significance is shown for survival probability among the samples.
Cytogenetic abnormalities in GATA2 deficiency. (A) Cytogenetic findings. Percentages of patients with the indicated cytogenetic abnormalities. The number of patients with trisomy 8 or monosomy 7 includes all patients with this abnormality, independent of other abnormalities. “Other,” abnormalities other than trisomy 8 or monosomy 7; “complex,” more than 1 abnormality in the same patient, including trisomy 8 and/or monosomy 7. (B) Cytogenetics vs age. The age distribution of cytogenetic findings, with the mean ages and standard deviations indicated (ANOVA, P = .1069). (C) Cytogenetics vs survival. Kaplan-Meier survival probability as a function of cytogenetics. Classifications are as in panel A. Survival is from the initial sample collection to the end of the study. (D) Diagnosis vs survival. Kaplan-Meier survival probability as a function of bone marrow diagnosis. Patients with AML or CMML were combined because of the low number of cases. Each graph shows the survival time from sample collection until the end point. Mantel-Cox test significance is shown for survival probability among the samples.
Cytogenetic abnormalities were rare in G2BMID (5%; 2 of 39) and common in MDS (68%; 32 of 47). However, because certain cytogenetic abnormalities define MDS in the setting of persistent cytopenia (eg, monosomy 7),17 the percentage is inherently higher in the MDS group than in G2BMID. Cytogenetic abnormalities were the most common in patients with AML or CMML (5 of 6). The presence of cytogenetic abnormalities was not biased by patient age (normal, 27.6 ± 12.8 years; abnormal, 31.4 ± 11.6 years; Student t test; P = .1586; Figure 2B). Monosomy 7 was more common in the male patients (25%; 8 of 33) vs the female patients (5%; 3/59; supplemental Figure 2A). All other cytogenetic abnormalities did not vary significantly between the sexes. Cytogenetic abnormalities did not significantly affect survival (P = .3637; Figure 2C).
Clinical diagnosis and survival
The survival rate for all patients with G2BMID, MDS, and AML/CMML was 82% (72 of 92), and the survival was 100% for asymptomatic patients (n = 14; Figure 2D). The asymptomatic group, consisting of family members of affected individuals, also lacked cytogenetic abnormalities (13 of 14 tested). Therefore, this group was considered separately in subsequent analyses of cytogenetic and mutational backgrounds. The survival probability for patients with G2BMID was not significantly higher than for those with MDS (P = .2119), but the AML/CMML group survival was significantly lower than all other groups (Figure 2D). Overall survival was not affected by sex or age (supplemental Figure 3B-C).
Somatic mutations
The acquisition of somatic mutations in oncogenic and tumor-suppressor genes is thought to drive leukemic transformation.11 Therefore, we used whole-exome and myeloid leukemia–based targeted gene array sequencing on peripheral blood and bone marrow samples to identify acquired somatic mutations. An average of 5.4 ± 4.0 filtered variants per sample were found among all samples by WES, with a range of 0 to 26 mutations per sample (Figure 3A; Table 1; supplemental Table 2). The patient in the AML/CMML group averaged a higher number of mutations (12.6 ± 8.6) than the other diagnoses (5.2 ± 3.2), but there was no significant difference among the other categories (analysis of variance [ANOVA], P = .2990). Gene mutations common in non-GATA2 MDS/AML, including TET2, EZH2, IDH1/2, RUNX1, TP53, and splicing factors (SF3B1, SRSF2, U2AF1, ZRSR2), were rare or absent in this cohort with GATA2 deficiency (Table 1).

Somatic mutations in patients with GATA2 deficiency. (A) Somatic mutations vs diagnosis. The total number of WES somatic variants per patient for each diagnosis category. The mean and standard deviation are shown. (B) Frequency of recurrent somatic mutations. The proportion of patients with the most frequently recurrent somatic mutations. (C) Recurrent mutations vs patient age. The age distribution of patients with each somatic mutation with the mean age and standard deviation indicated. (D) Recurrent mutations vs sex. The percent frequency of recurrent somatic mutations by patient sex. “None” indicates the lack of any recurrent somatic mutations. *Significant difference by ANOVA.
Somatic mutations in patients with GATA2 deficiency. (A) Somatic mutations vs diagnosis. The total number of WES somatic variants per patient for each diagnosis category. The mean and standard deviation are shown. (B) Frequency of recurrent somatic mutations. The proportion of patients with the most frequently recurrent somatic mutations. (C) Recurrent mutations vs patient age. The age distribution of patients with each somatic mutation with the mean age and standard deviation indicated. (D) Recurrent mutations vs sex. The percent frequency of recurrent somatic mutations by patient sex. “None” indicates the lack of any recurrent somatic mutations. *Significant difference by ANOVA.
ASXL1 and STAG2 were highly recurrent mutations (Figure 3B). ASXL1 mutations were found in 16% (n = 17 of 106) of all patients, with the most common mutation being c.1934_1935insG, p.646Wfs*12 (supplemental Table 2). STAG2 mutations were observed in 26% (n = 28) of the 106 patients; the location of these mutations was highly variable, but nearly all were truncation or splicing mutations. Mutations in DNMT3A (n = 12), BCOR (n = 4), and SETBP1 (n = 3) were also found. When pooled, mutations in the splicing factors U2AF1 and SRSF2 were found in 5 patients, but no mutations were found in SF3B1 or ZRSR2. Mutations in CUX1, KMT2A, RUNX1 and SMC1A were each found in two patients (Table 1). At least 1 cohesin- or chromatin-related gene mutation was found in ∼50% (54) of the 106 patients.
Age was not a significant factor in the occurrence of STAG2 or ASXL1 mutations (ANOVA; P = .0896; Figure 3C), although DNMT3A mutations occurred in older individuals than did mutations in ASXL1 or STAG2. However, ASXL1 mutations were highly enriched in female patients (23% of female vs 5% of male patients; Fisher’s exact test, P = .016), whereas STAG2 mutations were found more often in male patients (38% of male vs 20% female patients; Figure 3D). Abnormal cytogenetics were as frequent among the patients with either ASXL1 (47%; n = 8 of 17) or STAG2 (44%; 12 of 27) mutations, as they were in patients who lacked either of the mutated genes (Fisher’s exact test: ASXL1, P = .4216; STAG2, P = .4902). Neither mutated ASXL1 or STAG2 was present in asymptomatic patients but, critically, the mutations occurred similarly in patients with G2BMID or MDS (χ2 test, P = .5534; Table 1; Figure 4A). These data suggest that aberrant clones with malignancy-associated mutations are common in the premalignant G2BMID state. Patients with mutations in ASXL1, DNMT3A, or STAG2 all had lower probabilities of survival (Figure 4B). The total number of variants did not correlate with patients’ age, but 6 to 10 variants had a lower survival probability (Figure 4C). DNMT3A was found in 3 asymptomatic patients, and asymptomatic patient 23 had 4 common myeloid malignancy–associated mutations. The somatic mutations found in this cohort did not correlate with specific cytogenetic findings (Figure 4A).
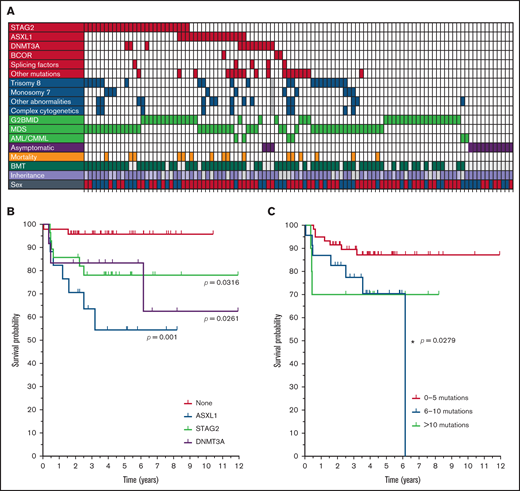
Somatic mutation occurrence and survival. (A) Summary of patients with GATA2 deficiency organized by somatic mutation status, cytogenetics, and diagnosis. Each vertical row represents 1 patient. Filled boxes indicate the presence of the parameter indicated on the left. Gray boxes indicate no data for that parameter. Recurrent mutations are shown in red, cytogenetic findings in blue, and diagnoses in green. Patients with STAG2 mutations are grouped together, followed by patients with mutations in ASXL1, then those with mutations in DNMT3A. Mutations in SRSF2 and U2AF1 are grouped together as “splicing,” and other common hematological mutations are grouped together as “other mutations” (Table 1). Aberrant cytogenetic findings are indicated as described in Figure 2. Familial inheritance is indicated by lavender boxes and de novo is pale purple. Males are indicated by blue and females by red. (B) Recurrent mutations and survival. Kaplan-Meier survival curves for the recurrent somatic mutations. Patients without a mutation in 1 of the recurrent genes are grouped together as “none.” The significance of each group compared with none is indicated. (C) Total somatic mutations and survival. Kaplan-Meier survival curves with the total number of somatic mutations determined by WES and grouped into 3 categories. Survival is from the initial sample collection. *Significant differences between groups.
Somatic mutation occurrence and survival. (A) Summary of patients with GATA2 deficiency organized by somatic mutation status, cytogenetics, and diagnosis. Each vertical row represents 1 patient. Filled boxes indicate the presence of the parameter indicated on the left. Gray boxes indicate no data for that parameter. Recurrent mutations are shown in red, cytogenetic findings in blue, and diagnoses in green. Patients with STAG2 mutations are grouped together, followed by patients with mutations in ASXL1, then those with mutations in DNMT3A. Mutations in SRSF2 and U2AF1 are grouped together as “splicing,” and other common hematological mutations are grouped together as “other mutations” (Table 1). Aberrant cytogenetic findings are indicated as described in Figure 2. Familial inheritance is indicated by lavender boxes and de novo is pale purple. Males are indicated by blue and females by red. (B) Recurrent mutations and survival. Kaplan-Meier survival curves for the recurrent somatic mutations. Patients without a mutation in 1 of the recurrent genes are grouped together as “none.” The significance of each group compared with none is indicated. (C) Total somatic mutations and survival. Kaplan-Meier survival curves with the total number of somatic mutations determined by WES and grouped into 3 categories. Survival is from the initial sample collection. *Significant differences between groups.
Discussion
GATA2 is a transcription factor central to maintaining the dynamic balance of hematopoiesis.18 In general, it promotes stem cell and myeloid precursor cell proliferation. A germline mutation in GATA2 leads to disruption of normal hematopoiesis and the loss of hematopoietic stem cells in the bone marrow, which may create a Darwinian selection gate for “breakaway” stem cells, in which acquired somatic mutations provide a proliferation and/or survival advantage. Consistent with this effect, patients with GATA2 deficiency can progress from hypocellular marrow, presumably putting stem cells under replicative stress, to hypercellular marrow, suggesting clonal hematopoiesis. Clonal hematopoiesis precedes myeloid malignancy, and mutations in ASXL1 and DNMT3A are common in patients with GATA2 deficiency and also in clonal hematopoiesis.19
This study was conducted to identify somatic mutations that lead to disease progression in patients with GATA2 deficiency/MonoMAC syndrome and to correlate these findings with biometric and other parameters. We recorded the patients’ age, sex, clinical features, cytogenetics and survival and compared the results with genetic profiles based on germline GATA2 mutations and acquired somatic mutations. Survival probabilities across the variables did not differ when patients who had undergone HCST were considered separately from patients who had not.
Several factors limit the ability of next-generation DNA sequencing to identify clinically important somatic mutations. Therefore, we used a conservative strategy to predict somatic mutations. The thresholds used in the analysis process were informed approximations, and the inclusion/exclusion boundaries were therefore inherently imprecise. The conservative estimations we used were meant to produce a plausible mutation list while minimizing the false positives that, once reported, tend to linger in databases. Our results are undoubtedly also affected by unknown patient enrollment biases, particularly patients evaluated for HSCT.
Somatic mutations in GATA2 were not identified in this study and are generally rare (1% to 5%) in myeloid malignancies, with the exception of biallelic mutations in CEBPA (CEBPAbi).20-26 In these cases, 18% to 40% of CEBPAbi-bearing patients have a somatic mutation in GATA2.27-31 However, CEBPA mutations were not detected in this cohort, indicating that CEBPAbi/GATA2-bearing patients are not among the patients with GATA2 deficiency in this study.
Patients with GATA2 deficiency typically present with symptoms of immunodeficiency with hypocellular marrows in adolescence and show signs of myeloid malignancy in early adulthood.8,10,32-35 However, we identified a distinct population of patients who do not develop symptoms of the disease at the typical age or progress into a diseased state as they age. These asymptomatic patients were identified by family screening after the proband tested positive for GATA2 mutation, constituted ∼13% of the cohort, and have been reported elsewhere at ∼10% to 30% of the studied cohort.6,8,33,36-38 In this and other studies, the asymptomatic group was the oldest, suggesting that a mutation in GATA2 alone is necessary but not sufficient for the development of disease. Indeed, the most critically ill patients are the youngest group. Asymptomatic individuals in this and other studies generally do not have the common somatic mutations associated with patients who have GATA2 deficiency with active disease or with non-GATA2 MDS/AML,6,37 although there was 1 striking exception. Three asymptomatic individuals with DNMT3A mutations were also exceptions. The mechanism underlying the incomplete penetrance of GATA2 mutations remains unknown. Among symptomatic patients, there is a trend for older patients to have MDS rather than G2BMID, as previously reported.6,37 In this study, the presence of somatic mutations was similar in both the G2BMID and MDS groups, as noted previously,6 indicating that clonal hematopoiesis is present at an early stage of immunodeficiency and cytopenia with hypoplastic marrow, even before the manifestation of MDS or myeloid malignancy. Moreover, the mutant allele frequencies (MAFs) for the common somatic mutations did not segregate by bone marrow diagnosis.
We determined the cytogenetics in patients with GATA2 deficiency, to identify relative risk factors for HSCT. We found that 44% of affected patients had abnormal cytogenetics, with trisomy 8 dominating (24%), followed by monosomy 7 (12%). The frequency of these anomalies has been reported, although in most other studies monosomy 7 was more common than trisomy 8.6,9,32,33,39-43 McReynolds and colleagues also reported trisomy 8 as the most common cytogenetic finding in a small cohort of patients with GATA2.6 A small subset of the patients in our study has been previously reported; however, our study is considerably larger and includes a broader range of disease. The overall frequency of abnormal cytogenetics is similar to non-GATA2 MDS/AML,44 but the frequency of trisomy 8 is notably higher in GATA2 deficiency. The der(1;7)(q10;p10) chromosomal abnormality was present in 5 patients in our study and in those with GATA2 deficiency in other studies.9,45 It is a rare myeloid abnormality usually associated with Shwachman-Diamond syndrome.46 The der(1;7)(q10;p10) frequently occurs with trisomy 8 in non-GATA2 MDS,47 and it was present in 3 of 5 cases in our study and in a patient with GATA2 deficiency in another study.45 It results in a 7q− genotype, but its inclusion with monosomy 7 did not change survival probability. In our studies, monosomy 7/7q− did not correlate with significantly lower survival probability, in contrast to non-GATA2 MDS. Complex cytogenetic findings were also more frequent in our study (∼11%) than in other studies.6,9,32,33,39-43
We investigated the acquisition of somatic mutations in patients with GATA2 deficiency to better understand and predict disease progression. Patients with GATA2 deficiency have a low number of unique somatic mutations compared with those with other myeloid malignancies. In particular, there is a conspicuous absence of the “usual suspects” of somatic mutations found in non-GATA2 MDS/AML, including the splicing factor SF3B1, and TET2, and IDH1/2. A recent study of 1809 patients with MDS showed TET2 to be the most common mutation (27%) followed by mutations in the splicing factor SF3B1 (23%).48 Other studies have found a very high frequency of splicing factor mutations (∼65%) and DNA methylation mutations (∼47%) in MDS.49,50 The lack of SF3B1 mutations is consistent with the absence of ringed sideroblasts among the patients with GATA2-deficient MDS. The lack of other common MDS/AML mutations in GATA2 deficiency, particularly TET2 and splicing factor mutations, has been found in other studies.6,9,37,38,43
The 2 significant exceptions to this trend are ASXL1 and STAG2. Mutations in ASXL1 are very common in all myeloid malignancies and are present in ∼20% of patients with GATA2 deficiency.21,22,24,26,44,50-52 A mutation in STAG2 is less common in hematological malignancies (2% to 8%),21,22,24,26,44,50,51,53,54 but it was the most common somatic mutation in patients with GATA2 deficiency (∼25%).
The ASXL1 mutations in non-GATA2 MDS/AML are found in older patients, with a significant bias toward males.55 However, the ASXL1 mutations in GATA2 deficiency did not show an age bias and were found far more frequently in female patients. We reported this trend in a previous study of a smaller patient population.13 ASXL1 mutations associate with TET2, U2AF1, RUNX1, SETBP1, STAG2, and EZH2 in non-GATA2 MDS/AML.22,48,49,55,56 However, we did not observe this association in patients with GATA2 deficiency. Some studies report that cohesin mutations are mutually exclusive with adverse cytogenetics,57 but we did not observe any significant correlation with mutations in either ASXL1 or STAG2 and abnormal cytogenetics. Pastor and colleagues found a high incidence of mutated ASXL1 concomitant with monosomy 7,43 but that was also not observed in our cohort. In our study, ASXL1 mutations had a negative effect on survival probability, as we reported previously.13 Mutated ASXL1 and cohesion mutations are also negative prognosticators across all myeloid malignancies.22,55,57
A high frequency of mutations in STAG2 is a particular feature of GATA2 deficiency compared with other hematological malignancies. STAG2 mutations have been reported in a small number of patients with GATA2 deficiency6,58,59 ; however, they now emerge as the leading somatic mutation in this disease. As seen in non-GATA2 MDS/AML and solid tumors,57 the STAG2 mutations in GATA2 deficiency were almost exclusively truncating mutations. Seven patients had 2 variant alleles of STAG2, with 6 of 7 of the patients being male. Given that STAG2 is on the X chromosome, 13 of 14 mutations are splice site or truncations, and the MAFs are very distinct, suggests 2 independent clonal populations in these patients. Curiously, the MAFs of the 2 STAG2 alleles were approximately the same in the female patient and in the male patient with a missense mutation, leaving the possibility of either 2 separate clones or compound heterozygous clones in these individuals. In non-GATA2–associated patients with MDS, STAG2 mutations have shown concurrence with ASXL1, RUNX1, and BCOR49,50,60 mutations, but this concurrence was not observed in our study. McReynolds and colleagues found STAG2 solely in MDS patients6 ; however, we found both ASXL1 and STAG2 mutations in similar frequencies in patients with G2BMID and those with MDS.
STAG2 mutations are also very common in myeloid leukemia–Down syndrome.61,62 STAG2 was the most common cohesin mutation in our cohort, but there were also 1 RAD21 and 2 SMC1A mutations. These other cohesin gene mutations were more frequent in patients with myeloid leukemia–Down syndrome and included RAD21, SMC1A, and also SMC3 and CTCF. These 2 groups of patients also share a conspicuous lack of the most common MDS/AML mutations, including those in TET2, IDH1/2, and SF3B1.
The other less frequently recurring mutations include DNMT3A, BCOR, and SETBP1; the splicing genes SRSF2 and U2AF1; and the histone modifiers KMT2A and KDM2A. DNMTA3, BCOR, and SETBP1 have been reported in patients with GATA2 deficiency, along with NRAS and RUNX1.6,9,38,40,43,63-66
In summary, GATA2 deficiency is characterized by a novel pattern of somatic mutations with a strong bias toward chromatin- and cohesin-related genes and a notable absence of nearly all other common MDS/AML-associated somatic mutations. The 3 most common mutations are STAG2, ASXL1, and DNMT3A. The significance of mutations in ASXL1 and STAG2 on the GATA2 mutation background and their relevance to myeloid transformation remain a topic for future investigation.
Acknowledgments
The authors thank the National Cancer Institute (NCI) Center for Cancer Research (CCR) Genomics Core for their assistance in direct and array DNA sequencing.
This work was supported, in whole or (in part), by federal funds from NCI, National Institutes of Health (NIH; HHSN261200800001E; ZIABC010870). The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. This research was supported in part by the Intramural Research Program of the NIH, NCI, CCR, and the National Institute of Allergy and Infectious Diseases and the NIH Clinical Center Research Award for Staff Clinicians (RASCL) program.
Authorship
Contribution: R.R.W. designed the research, performed the experiments and data analysis, and drafted the manuscript; K.R.C. performed the experiments, provided critical revision of the manuscript for important intellectual content, and reviewed the histopathology; L.J.E. and W.W. performed the experiments and data analysis; L.M.T. performed the experiments; T.R.B. and D.T. performed the data analysis and provided critical revision of the manuscript for important intellectual content; J.L. performed data analysis; S.D. performed the experiments; A.P.H and S.M.H. provided critical revision of the manuscript for important intellectual content; and D.D.H. supervised the study and drafted the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Dennis D. Hickstein, National Cancer Institute, National Institutes of Health, 9000 Rockville Pike, Bldg 10/CRC, Room 3-3142, Bethesda, MD 20892; e-mail: hicksted@mail.nih.gov.

