

TO THE EDITOR:
Mantle cell lymphoma (MCL) is molecularly dependent on B-cell receptor (BCR) signaling.1 The Bruton tyrosine kinase (BTK) inhibitor ibrutinib suppresses a key mediator of the BCR pathway, with response rates of 67% to 77% in patients with relapsed or refractory (R/R) MCL.2,3 However, the depth of responses is limited, with complete remission (CR) rates of 21% to 38%, and BTK resistance frequently occurs, with a median progression-free survival (PFS) of less than18 months.2-5
Preventing resistance may require inhibition of alternative pathways, such as the PI3K pathway.6,7 In vitro studies combining PI3K and BTK inhibitors demonstrate synergistic cytotoxicity.8 In particular, inhibition of multiple PI3K isoforms, including α and δ, prevented resistance in B-cell lymphomas, including an MCL model.9-11
Recent studies combining PI3K–δ-specific inhibitors with BTK inhibitors, including umbralisib/ibrutinib and idelalisib/tirabrutinib, showed a tolerable toxicity profile, but efficacy was not improved compared with historical studies of ibrutinib alone, possibly due to the narrow activity of PI3K–δ-specific inhibitors.12-15 Combining the pan-PI3K inhibitor buparlisib with ibrutinib demonstrated a promising complete response rate, but additive toxicities prevented further development.16
Copanlisib, a pan-PI3K inhibitor with specificity for α and δ isoforms, has established single-agent activity in MCL, with response rates of 64% and a favorable toxicity profile.17 We hypothesized that the combination of copanlisib with ibrutinib would improve the rate and durability of response to treatment in patients with MCL.
We performed a phase 1 study evaluating combination copanlisib and ibrutinib in patients with R/R MCL. Patients with R/R MCL after ≥1 prior line of therapy, good performance status, and adequate bone marrow and organ function were eligible for enrollment. Previous treatment with either BTK or PI3K inhibitor was permitted if the best response to BTK or PI3K inhibitor was stable disease or better. This phase 1 clinical trial was performed in accordance with its design as approved by the Institutional Review Board at Memorial Sloan-Kettering Cancer Center and performed in accordance with the Declaration of Helsinki. It is registered at www.clinicaltrials.gov as NCT03877055.
Copanlisib was given by IV on days 1, 8, and 15 of 28-day cycles. Ibrutinib was given orally every day in 28-day cycles. Dose escalation occurred in a 3 + 3 scheme (dose level 1, copanlisib 45 mg/ibrutinib 560 mg; dose level 2, copanlisib 60 mg/ibrutinib 560 mg). The maximum duration of treatment was 36 cycles, not exceeding 36 months. In patients achieving a CR by RECIL (response evaluation criteria in lymphoma) criteria, copanlisib was held after 2 additional cycles, and ibrutinib monotherapy was continued. Safety was assessed at screening and throughout the duration of the trial through assessments, including physical examination, electrocardiograms, clinical laboratory testing, and vital sign assessments. Efficacy was evaluated via fluorodeoxyglucose-positron emission tomography/computed tomography (FDG-PET/CT) or CT at regular intervals starting in cycle 3.
The primary aim was the identification of the maximum tolerated dose (MTD) and recommended phase 2 dose. Secondary aims included identifying the complete response rate (CRR) within 6 months, calculated via Simon 2-stage mini-max design with a 1-sided type I error rate of 0.05 and power (1 - β) of 0.80. Other secondary objectives, including overall response rate (ORR), PFS, and event-free survival, were classified according to International Working Group criteria and calculated using Kaplan-Meier estimation assessment for minimal residual disease (MRD), which was performed using next-generation sequencing (NGS) of immunoglobulin rearrangements using clonoSEQ.18-20
From March 2019 through October 2020, 8 patients were enrolled (dose level 1, 6 patients; dose level 2, 2 patients) (Table 1). All patients had received 1 prior line of therapy, including 1 patient who received ibrutinib. This patient was treated with front-line bendamustine, rituximab, and ibrutinib, achieved CR, and had disease relapse while on maintenance ibrutinib before enrollment. High-risk pathologic features at enrollment included blastoid or pleomorphic histology in 62.5%, elevated Ki-67 proliferation index in 75%, and TP53 aberrations in 87.5%. TP53 aberrancies were noted in 5 of 7 patients at initial diagnosis and 2 at repeat biopsy before enrollment.
Baseline patient characteristics
| Characteristic | n = 8, n (%) |
|---|---|
| Histology | |
| MCL | 8 (100) |
| Age, median (IQR) | 67.0 (62.8-71.0) |
| Sex | |
| Female | 2 (25) |
| Male | 6 (75) |
| Stage | |
| 3 | 1 (12) |
| 4 | 7 (88) |
| Prior lines of therapy | |
| 1 | 8 (100) |
| Prior ibrutinib | 1 (12) |
| Prior stem cell transplant | 0 (0) |
| KI 67 at enrollment | |
| <30% | 2 (25) |
| ≥30% | 6 (75) |
| Histology | |
| MCL, NOS | 3 (38) |
| MCL, blastoid variant | 4 (50) |
| MCL, pleomorphic variant | 1 (12) |
| LDH at enrollment above ULN | 4 (50) |
| Lesion >5 cm | 3 (38) |
| Number of extranodal sites | |
| <2 | 4 (50) |
| ≥2 | 4 (50) |
| Baseline SUV max, median (IQR) | 14 (8-26) |
| TP53 deletion at enrollment* | 3 (38) |
| TP53 mutation at enrollment* | 5 (62) |
| Characteristic | n = 8, n (%) |
|---|---|
| Histology | |
| MCL | 8 (100) |
| Age, median (IQR) | 67.0 (62.8-71.0) |
| Sex | |
| Female | 2 (25) |
| Male | 6 (75) |
| Stage | |
| 3 | 1 (12) |
| 4 | 7 (88) |
| Prior lines of therapy | |
| 1 | 8 (100) |
| Prior ibrutinib | 1 (12) |
| Prior stem cell transplant | 0 (0) |
| KI 67 at enrollment | |
| <30% | 2 (25) |
| ≥30% | 6 (75) |
| Histology | |
| MCL, NOS | 3 (38) |
| MCL, blastoid variant | 4 (50) |
| MCL, pleomorphic variant | 1 (12) |
| LDH at enrollment above ULN | 4 (50) |
| Lesion >5 cm | 3 (38) |
| Number of extranodal sites | |
| <2 | 4 (50) |
| ≥2 | 4 (50) |
| Baseline SUV max, median (IQR) | 14 (8-26) |
| TP53 deletion at enrollment* | 3 (38) |
| TP53 mutation at enrollment* | 5 (62) |
LDH, lactate dehydrogenase; NOS, not otherwise specified; SUV, standardized uptake value.
One patient's MCL had both a TP53 mutation and TP53 deletion.
The MTD was dose level 1 (copanlisib 45 mg/ibrutinib 560 mg). Two patients enrolled at dose level 2 experienced dose-limiting toxicities (DLT). DLTs included grade 3 acute kidney injury requiring temporary interruption of ibrutinib and grade 3 rash requiring oral steroids and interruption of copanlisib and ibrutinib, with both drugs resumed at reduced doses. Given these DLTs, dose level 1 was expanded and selected as the MTD. At dose level 1, 1 patient had grade 3 mucositis requiring drug interruption and resumed treatment at a lower dose with improvement in symptoms. Grade 3 or 4 adverse events (AEs) occurring in ≥2 patients included grade 3 rash in 3 patients and transient grade 3 diarrhea in 2 patients. One patient discontinued therapy due to hepatotoxicity after 50 days of treatment; they subsequently resumed ibrutinib monotherapy with no further hepatotoxicity noted. Three patients had asymptomatic cytomegalovirus (CMV) reactivation on surveillance polymerase chain reaction not requiring treatment and spontaneous reversion to CMV negativity. A full list of treatment-related AEs is summarized in supplemental Table 1.
With a median follow-up of 20 months (range, 8-20 months), the median time on both copanlisib and ibrutinib was 111 days (range, 50-350 days). All 8 patients were evaluable for efficacy. ORR was 87.5% with CR in 4 patients (50%) and PR in 3 patients (37.5%). The only patient treated with ibrutinib before trial enrollment had progressive disease on trial as best response. Median PFS was 7.7 months (95% confidence interval, 2.7-NR) (Figure 1). Two patients proceeded to chimeric antigen receptor T-cell therapy and 1 patient to allogeneic transplant after disease progression on copanlisib/ibrutinib.
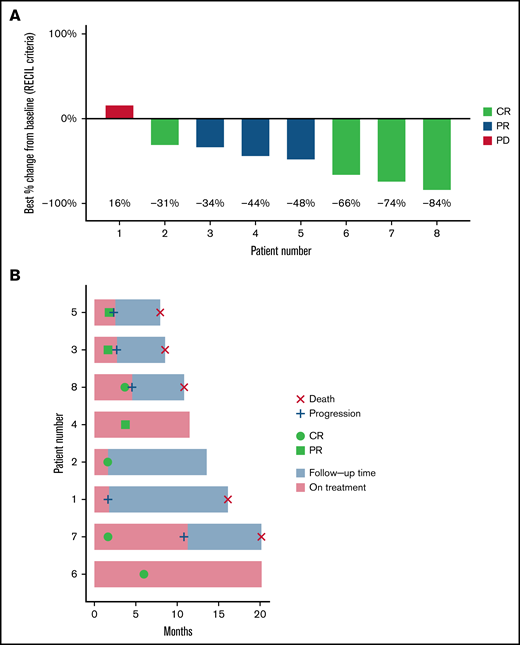
Response to treatment with combination copanlisib/ibrutinib. (A) Waterfall plot of best responses demonstrating 87.5% response rate. Percent change in index lesions is reflected in the y-axis, while categorical response by RECIL criteria, which incorporates both size and FDG avidity, is reflected by color index. Patient 2 had CR by RECIL criteria based on a decrease in the sum of the longest diameters of index lesions of at least 30%, plus normalization of FDG-PET avidity. (B) Swimmer plot of all 8 treated patients.
Response to treatment with combination copanlisib/ibrutinib. (A) Waterfall plot of best responses demonstrating 87.5% response rate. Percent change in index lesions is reflected in the y-axis, while categorical response by RECIL criteria, which incorporates both size and FDG avidity, is reflected by color index. Patient 2 had CR by RECIL criteria based on a decrease in the sum of the longest diameters of index lesions of at least 30%, plus normalization of FDG-PET avidity. (B) Swimmer plot of all 8 treated patients.
MRD assessment by NGS demonstrated detectable residual disease in 7 of 8 patients, including detectable disease in all 4 patients with radiologic CR by RECIL criteria. One patient in radiologic partial response had test failure due to a low number of B cells in the sample, possibly consistent with undetectable disease.
There were no treatment-related deaths. Five patients had progression of disease on clinical trial and subsequently died due to complications from MCL progression. The other 3 patients are alive at the time of publication, with 2 continuing treatment per protocol.
A phase 2 cohort was initially planned with a target accrual of 33 patients; however, due to poor accrual, toxicity, and the shifting treatment landscape for MCL, the study was closed early.
Our study illustrates clinical outcomes with the PI3K α/δ inhibitor copanlisib and the BTK inhibitor ibrutinib in R/R MCL. This combination was designed to inhibit BCR signaling more thoroughly than ibrutinib alone, with the hope of achieving better outcomes in R/R MCL. While a high ORR and complete response rate were observed in this small cohort, responses were of limited duration, and the regimen was further limited by additive toxicity.
Establishing the value of adding copanlisib to ibrutinib is difficult given the small sample size and the unique population itself. All patients had only received 1 prior line of therapy, though they represented a particularly high-risk population, with 87.5% of patients exhibiting TP53 aberrant disease and 75% with Ki-67 over 30%. The ORR of 87.5% with a CR rate of 50% compares favorably with ibrutinib monotherapy, though responses were short-lived, with 50% of patients experiencing progression of disease on treatment ≤6 months.2-5 Limitations to the dose intensity of copanlisib and ibrutinib, and early cessation of copanlisib per study design, may have contributed to the short response times. Interestingly, MRD assessment by NGS remained detectable in all but 1 patient in this study, reflecting the limited durability of responses and highlighting the potential for MRD assessment as a predictor of PFS and OS in MCL.21
A similar study of the oral pan-PI3K inhibitor buparlisib in combination with ibrutinib revealed promising efficacy with a 94% response rate in MCL patients, but this regimen was not pursued due to buparlisib-associated neuropsychiatric toxicity, underlining the challenges in combined targeted therapies for lymphoma.15,16 While copanlisib/ibrutinib did not cause neurotoxicity, the AE profile and logistical challenges to administration precluded further development of this regimen. More effectively inhibiting critical signaling pathways in MCL while mitigating the on-target, off-tumor toxicities of combination agents will require further investigation with alternative drugs and targets. Other combination targeted studies are ongoing, such as a trial of the BTK inhibitor zanubrutinib combined with a PI3K inhibitor, zandelisib, which will be administered on an interrupted dosing schedule which may mitigate toxicity (NCT02914938). These future studies may provide additional insights and improve the efficacy and tolerability of combined targeted therapy in MCL.
Acknowledgments: The authors thank the investigators and patients of this trial for their participation and the clinical trial teams, including Ashley Ames, Shelley Levi, and Joanna Dicostanza, for assistance in managing patients and managing regulatory and data aspects of the clinical trial. Our clinical trials would not be able to function without the dedicated support of our clinical research team members.
Supported by Memorial Sloan-Kettering Cancer Center (MSK), with additional monetary and therapeutic drug support from Bayer. This research was also funded in part by core grants as part of MSK Lymphoma SPORE (P50 CA192937), NIH/NCI Cancer Center Support Grant (P30 CA008748) to MSK. The work of C.B. has been supported by an ASCO Young Investigator Award, an ASH Clinical Scholar Award, and the Lymphoma Research Foundation Clinical Research Mentoring Program. The authors also thank the George L. Ohrstrom Foundation, Steven A. Greenberg Award for support of our clinical translational research. The authors would like to acknowledge the contributions of the Bayer research team, including Kelli Janousek, Dereck Amakye, Sanjeeva Reddy, Dennis Healy, and Barry Childs, for discussions of clinical trial concept and guidance throughout the study.
Contribution: D.Q. and C.B. collected and analyzed data and wrote the manuscript; H.Y.L., J.A.E., S.S., and E.B. oversaw the execution and logistics of the clinical trial and provided data generation; K.W. and V.S. performed biostatistical analysis; A.K., M.M., C.O., and C.B. enrolled and oversaw care of patients on the clinical trial; A.Y. and C.B. designed the study; and G.S. contributed to data analysis and reviewed and edited the manuscript.
Conflict-of-interest disclosure: A.K. receives research support from AbbVie, Adaptive Biotechnologies, Celgene, Pharmacyclics, and Seattle Genetics; and has an advisory role with Celgene. M.M. receives research support from Genentech, Roche, GlaxoSmithKline, Bayer, Pharmacyclics, Janssen, Rocket Medical, and Seattle Genetics; has received honoraria from Genentech, Roche, Bayer, Pharmacyclics, Janssen, Seattle Genetics, and GlaxoSmithKline; and has a consultancy role with Genentech, Bayer, Merck, Juno, Roche, Teva, Rocket Medical, and Seattle Genetics. G.S. has a consultancy role with Abbvie, Bayer, Beigene, BMS/Celgene, Debiopharm, Epizyme, Genentech/Roche, Genmab, Incyte, Ipsen, Janssen, Kite/Gilead, Loxo, Milteniy, Morphosys, Novartis, Rapt, Regeneron, Takeda, and Velosbio. A.Y. is employed at AstraZeneca. C.L.B. receives research support from Janssen, Novartis, Epizyme, Autolus, Roche, and Bayer; has received honoraria from Dava Oncology; and has a consultancy role with Skipta, Kite Pharma, MorphoSys, Bristol-Myers Squibb, Karyopharm Therapeutics, Genentech, and TG Therapeutics. All other authors declare no competing financial interests.
Correspondence: Connie Batlevi, Lymphoma Service, Department of Medicine, Memorial Sloan-Kettering Cancer Center, 1275 York Avenue, New York, NY, 10065; e-mail: leec@mskcc.org.

