


TO THE EDITOR:
MECOM (MDS1 and EVI1 complex locus, MIM: 165215) encodes the transcriptional regulators EVI1 and MDS1-EVI1, which are transcribed from alternative transcriptional start sites.1 In normal human cells, EVI1 plays important roles in hematopoiesis and stem cell self-renewal.2 The overexpression of EVI1 and MDS1-EVI1 has been reported in various malignancies, such as acute myeloid leukemia and solid tumors.2,3 MDS1 has a PRDI-BF1-RIZ1 domain with histone methyltransferase activity, and EVI1 possesses an N-terminal zinc finger (ZF) domain, which comprises 7 C2H2 ZF motifs and a C-terminal ZF domain with 3 C2H2 ZF motifs (Figure 1A).4
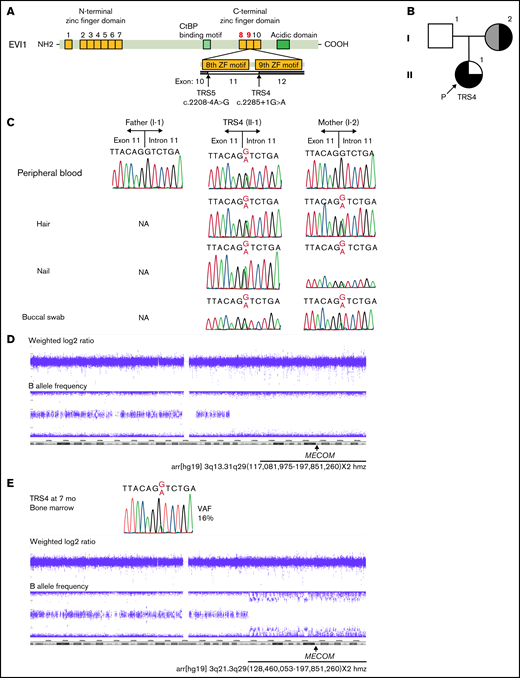
Analysis of MECOM in TRS4 and her family. (A) Graphical representation of the EVI1 protein and its major functional domains. EVI1 harbors 7 ZF motifs in the N-terminal ZF domain and 3 ZF motifs in the C-terminal ZF domain. Arrows indicate the exon boundaries affected by the variants in this study. (B) Pedigree of family 1. Black on the left represents pancytopenia, gray on the left represents mild leukocytopenia and thrombocytopenia, black on the upper right represents radioulnar synostosis, and black on the lower right represents clubfoot. P indicates the proband (TRS4). (C) Mutation analysis of TRS4 and her family. The genomic DNA sources are listed on the left. (D) The results of SNP array using the DNA from leukocytes in the peripheral blood of the mother of TRS4. The weighted log 2 ratio revealed neutral copy number, and the B allele frequency showed the loss of heterozygosity from 3q13.31 to 3q29. (E) The results of Sanger sequencing and SNP array obtained using the DNA from the bone marrow of TRS4 at 7 months of age. The mutant A peak was smaller than the G (wild-type) peak in the electropherogram. SNP array showed copy-neutral loss of heterozygosity from 3q21 to 3q29. NA, not available; SNP, single nucleotide polymorphism; VAF, variant allele frequency determined by WES.
Analysis of MECOM in TRS4 and her family. (A) Graphical representation of the EVI1 protein and its major functional domains. EVI1 harbors 7 ZF motifs in the N-terminal ZF domain and 3 ZF motifs in the C-terminal ZF domain. Arrows indicate the exon boundaries affected by the variants in this study. (B) Pedigree of family 1. Black on the left represents pancytopenia, gray on the left represents mild leukocytopenia and thrombocytopenia, black on the upper right represents radioulnar synostosis, and black on the lower right represents clubfoot. P indicates the proband (TRS4). (C) Mutation analysis of TRS4 and her family. The genomic DNA sources are listed on the left. (D) The results of SNP array using the DNA from leukocytes in the peripheral blood of the mother of TRS4. The weighted log 2 ratio revealed neutral copy number, and the B allele frequency showed the loss of heterozygosity from 3q13.31 to 3q29. (E) The results of Sanger sequencing and SNP array obtained using the DNA from the bone marrow of TRS4 at 7 months of age. The mutant A peak was smaller than the G (wild-type) peak in the electropherogram. SNP array showed copy-neutral loss of heterozygosity from 3q21 to 3q29. NA, not available; SNP, single nucleotide polymorphism; VAF, variant allele frequency determined by WES.
Recently, we reported missense mutations at the eighth ZF motif in MECOM as one of the causes of radioulnar synostosis (RUS) with amegakaryocytic thrombocytopenia (RUSAT, MIM: 616738), which is characterized by bone marrow failure and congenital proximal fusion of the radius and ulna, which ultimately results in extremely limited pronation and supination of the forearm.5 Clinodactyly, clubfoot, neurosensory hearing impairments, tetralogy of Fallot, and nail abnormalities are also manifested in individuals with variants in MECOM.5-8 Additional studies have revealed that nonsense, frameshift, and splicing (supposed to be loss of function) variants in MECOM also cause bone marrow failure but not RUS.6-8 In contrast, individuals with missense variants at the eighth and ninth ZF motifs present diverse hematological and skeletal defects, including RUS (supplemental Figure 1).5-13 However, the mechanisms underlying the heterogeneity of hematological symptoms have not been explored. Here, we identified 2 novel splicing variants in 2 individuals with bone marrow failure as well as in their families. In one family, we found reduced variant fractions due to copy neutral loss of heterozygosity (CNLOH). This study was approved by the Ethics Committee of Tohoku University School of Medicine, which was performed in accordance with the Declaration of Helsinki.
An infant girl, TRS4, was the first child of her Japanese parents (Figure 1B; supplemental Notes). Her father was healthy, whereas her mother had bilateral RUS and mild leukocytopenia and thrombocytopenia. Immediately after birth, she was found to have macrocytic anemia (hemoglobin 5.8 g/dL, mean corpuscular volume 140.9 fl, mean corpuscular hemoglobin concentration 33.0%) and thrombocytopenia (platelet count 9000/μL), and she presented with bilateral clubfeet but no RUS. Sanger sequencing of MECOM in the DNA retrieved from leukocytes in the peripheral blood of TRS4 at 1 month of age was performed, and a novel splicing variant was identified (NM_001105078:c.2285 + 1G>A, Figure 1C; supplemental Figure 1A). The variant was not identified in the DNA from leukocytes in the peripheral blood of her parents. Whole-exome sequencing (WES) of DNA from the mother also did not identify the variant (0/88 total reads). However, the SNP array of DNA from the mother revealed CNLOH in 3q13.31q29, where MECOM is located (Figure 1D). We identified the splicing variant in the DNA from a buccal swab as well as the hair and nail samples of the mother (Figure 1C). We also confirmed that this variant resulted in in-frame exon skipping (NM_001105078:r.2460_2537del, NP_001098548:p.Tyr736_Arg762del) and retained intron, leading to premature termination (NP_001098548:p.Arg762_Cys763insSerAspAsnAspIleCysTer, supplemental Figures 1A-B and 2A-C). These results suggest that somatic genetic event occurred in the leukocytes of the mother of TRS4.
Subsequently, TRS4 was relieved from transfusion-dependent anemia despite being dependent on repetitive platelet transfusion. At 7 months of age, we performed Sanger sequencing of the DNA from the bone marrow of TRS4. We found that the fraction of the variant was lower than that of leukocytes at 1 month of age (Figure 1E). To count the variant allele frequency (VAF), WES was performed on DNA from the bone marrow of TRS4, and a VAF of 16% (12/76 reads in WES) was obtained. SNP array analysis revealed CNLOH in 3q21.3q29, including MECOM (Figure 1E). Thereafter, TRS4 gradually developed anemia, and her bone marrow was reexamined at 11 months of age. The VAF in the bone marrow and leukocytes in the peripheral blood were 23% (25/107 reads) and 42% (30/72 reads), respectively. Consequently, TRS4 became dependent on once-a-week transfusion. An unrelated bone marrow transplantation was also conducted at 14 months of age.
A girl, TRS5, was the second child of her Japanese parents (Figure 2A; supplemental Notes). Her mother was healthy, whereas her father and uncle, who were monozygotic twins, had RUS, with no hematological abnormalities (Figure 2B). An older brother of TRS5 had bilateral clinodactyly of the fifth digit and a normal blood cell count. TRS5 was found to develop petechiae immediately after birth. Blood cell count revealed that she had anemia (hemoglobin 7.3 g/dL) and thrombocytopenia (platelet count 4000/μL). In addition, she had bilateral clinodactyly without RUS. Cord blood transplantation (CBT) was conducted when she was aged 8 months. Sanger sequencing of MECOM in the DNA from leukocytes in the peripheral blood before CBT revealed a novel splicing variant (NM_001105078:c.2208-4A>G, Figure 2C; supplemental Figure 1A) in TRS5. The father and brother of TRS5 also possessed this variant. RNA analysis of the peripheral blood leukocytes of TRS5 confirmed a 3-base insertion (NM_001105078:r.2459_2460insCAG), which was predicted to be 1 amino acid insertion (NP_001098548:p.Cys735_Arg736insSer, Figure 2D-E; supplemental Figure 1B). We confirmed the expression of the same insertion in the peripheral blood leukocytes of the father and brother of TRS5 (Figure 2C). These results suggest that the hematological phenotypes in family 2 were independent of the expression of the mutant transcript in leukocytes.
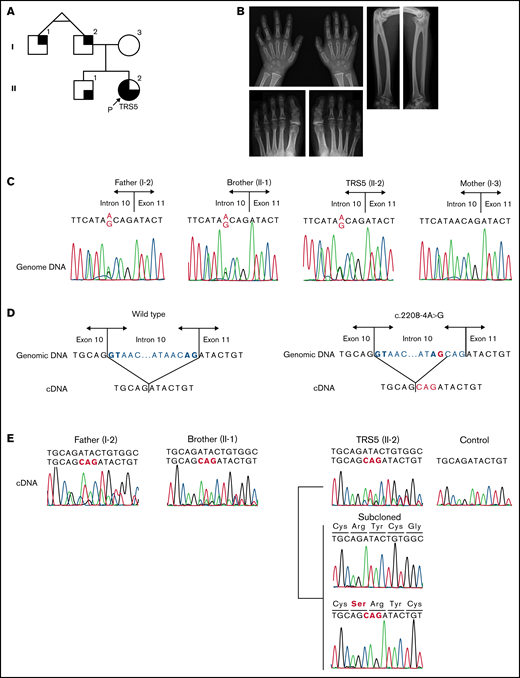
Analysis of MECOM in TRS5 and her family. (A) Pedigree of family 2. Black on the left represents pancytopenia, black on the upper right represents radioulnar synostosis, and black on the lower right represents clinodactyly of the fifth finger of hands. P indicates the proband (TRS5). (B) Radiographs of the individuals with c.2208-4A>G variants. Left, bilateral clinodactyly of the fifth fingers of the brother of TRS5 (II-1). Lower left, PIP joint fusion of bilateral second toes were suspected. Fusions of the DIP joint of the fourth and fifth toes on the left foot and third, fourth, and fifth toes on the right foot of the father of TRS5 (I-2). The incidence of DIP fusion in the Japanese population is 72.5% in the fifth toe, 11.9% in the fourth toe, and 0.8% in the third toe.17 Right, bilateral radioulnar synostosis in the father of TRS5 (I-2). (C) The c.2208-4A>G variant in MECOM in TRS5 and her family. The variant was identified in TRS5 as well as her brother and father. (D) A schema showing the splicing of wild-type (left) and mutant (right) RNA. Bold letters show GT-AG consensus nucleotides. (E) cDNA analysis revealed the presence of the in-frame insertion in TRS5 as well as her father and brother. PCR primer set 2 was used for TRS5 and her brother, and PCR primer set 3 was used for her father. cDNA, complementary DNA; DIP, distal interphalangeal; PCR, polymerase chain reaction; PIP, proximal interphalangeal.
Analysis of MECOM in TRS5 and her family. (A) Pedigree of family 2. Black on the left represents pancytopenia, black on the upper right represents radioulnar synostosis, and black on the lower right represents clinodactyly of the fifth finger of hands. P indicates the proband (TRS5). (B) Radiographs of the individuals with c.2208-4A>G variants. Left, bilateral clinodactyly of the fifth fingers of the brother of TRS5 (II-1). Lower left, PIP joint fusion of bilateral second toes were suspected. Fusions of the DIP joint of the fourth and fifth toes on the left foot and third, fourth, and fifth toes on the right foot of the father of TRS5 (I-2). The incidence of DIP fusion in the Japanese population is 72.5% in the fifth toe, 11.9% in the fourth toe, and 0.8% in the third toe.17 Right, bilateral radioulnar synostosis in the father of TRS5 (I-2). (C) The c.2208-4A>G variant in MECOM in TRS5 and her family. The variant was identified in TRS5 as well as her brother and father. (D) A schema showing the splicing of wild-type (left) and mutant (right) RNA. Bold letters show GT-AG consensus nucleotides. (E) cDNA analysis revealed the presence of the in-frame insertion in TRS5 as well as her father and brother. PCR primer set 2 was used for TRS5 and her brother, and PCR primer set 3 was used for her father. cDNA, complementary DNA; DIP, distal interphalangeal; PCR, polymerase chain reaction; PIP, proximal interphalangeal.
In the present study, we identified 2 novel variants in MECOM in subjects from 2 families that had a partially overlapping phenotype with RUSAT. These variants were classified as likely pathogenic according to the guidelines of American College of Medical Genetics and Genomics.14 In addition, the results of functional analysis suggested that these variants induced transcriptional dysregulation, similar to that caused by missense variants at the eighth ZF motif (supplemental Figure 3). Genetic testing of MECOM should be considered for individuals with RUSAT as well as those with congenital bone marrow failure and a family history of RUS. Detection of genetic variants and somatic genetic events will provide valuable knowledge for genetic counseling.
The cause of the phenotypic differences between TRS4 and her mother, who also had CNLOH, was largely unclear. The allele frequency of somatic variant is affected by the fitness of cells harboring the variant, genetic drift, and the time in life when the variant emerges. The vast majority of detectable clones are selected through positive fitness advantage, not drift, in clonal hematopoiesis.15 The fitness advantage of cells with the recovered allele could be associated with the phenotype of the mother. By contrast, TRS4 required hematopoietic stem cell (HSC) transplantation. A possible explanation for the failure of hematopoietic recovery in TRS4 is stem cell exhaustion; telomere length (TL) shortening may limit the replicative capacity of the HSCs.16 We observed marked TL shortening by 11 months of age in both leukocytes and bone marrow cells from TRS4 (supplemental Table 1). This TL shortening in TRS4 may be due to the rapid division of stem/progenitor cells with CNLOH. However, the TL attrition alone could not explain the phenotypic difference between TRS4 and her mother. Other factors, including genetic background and the time in life when CNLOH occurred, may affect the phenotype.
Further studies are warranted to elucidate the frequency and mechanisms of genetic reversion in bone marrow failure in subjects with MECOM-associated syndrome.
Acknowledgments: The authors would like to thank the patients, their families, and doctors who participated in the present study. They are grateful to Joan Massague and Jeff Wrana for providing p3TP-lux (Addgene plasmid #11767). They would also like to thank Yoko Tateda, Kumi Kato, Riyo Takahashi, Mami Kikuchi, and Kiyotaka Kuroda for their technical assistance.
Funding was provided by JSPS KAKENHI grants JP17K10045 and JP20H03637, Takeda Science Foundation, and the Japan Foundation for Pediatric Research.
Contribution: T.N., T.A., Y.O., N.I., and Y.A. performed and interpreted the Sanger sequencing and luciferase analysis data; R.T., Y.S., A.S., M. Irie, Y.S.N., S.K., and M. Imaizumi collected samples and clinical data of participants in this study; R.F., M.S., and K.N. performed whole-exome sequencing; and T.N., R.T., and Y.A. wrote and edited the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Tetsuya Niihori, Department of Medical Genetics, Tohoku University School of Medicine, 1-1 Seiryo-machi, Sendai 980-8574, Japan; e-mail: tniihori@med.tohoku.ac.jp.

