

Abstract
Although CAR T-cell therapy is US Food and Drug Administration–approved for B-cell non-Hodgkin lymphomas, the development of adoptive immunotherapy for the treatment of classic Hodgkin lymphoma (cHL) has not accelerated at a similar pace. Adoptive T-cell therapy with Epstein-Barr virus–specific cytotoxic T lymphocytes and CD30 CAR T cells have demonstrated significant clinical responses in early clinical trials of patients with cHL. Additionally, CD19 and CD123 CAR T cells that target the immunosuppressive tumor microenvironment in cHL have also been investigated. Here we discuss the landscape of clinical trials of adoptive immunotherapy for patients with cHL with a view toward current challenges and novel strategies to improve the development of CAR T-cell therapy for cHL.
Introduction
Classic Hodgkin lymphoma (cHL) is a unique lymphoid neoplasm that is characterized by malignant Hodgkin Reed-Sternberg (HRS) cells surrounded by a heterogenous mixture of nonmalignant inflammatory cells. With the advancement in treatments over the last few decades, most patients who develop cHL are cured with frontline therapy. However, about 10% to 15% of patients with early-stage and 15% to 30% of those with advanced-stage cHL will not be cured with frontline therapy.1-3 Autologous stem cell transplant offers an approximately 50% cure rate for those with relapsed or refractory (R/R) disease,4 and recent advances in treatment with the use of brentuximab vedotin (BV) and programmed cell death-1 (PD-1) checkpoint inhibitors have also expanded therapeutic options.5,6 However, treatment of those who progress after transplant or are ineligible for transplant still poses a significant challenge and thus requires novel approaches.
In cHL patients, T cells that could potentially recognize neoplastic cells are otherwise inhibited by several factors, including immunosuppressive cytokines, regulatory T cells, tumor-associated macrophages (TAMs), and the expression of checkpoint proteins (Figure 1). Adoptive T-cell immunotherapy has the ability to harness the power of cytotoxic T cells to circumvent these tumor-evasion strategies.7 With adoptive T-cell immunotherapy, autologous T cells are harvested from patients, expanded and engineered ex vivo, then reinfused back into the patient where they find cancer cells, kill them, and proliferate. Additionally, allogeneic T cells collected from healthy donors are being investigated as well. Chimeric antigen receptor (CAR) T-cell therapy is a form of adoptive immunotherapy that uses T cells and genetically engineers them to express CARs that redirect T-cell specificity to target tumor antigens.8 CARs are proteins that are comprised of an extracellular antigen-binding domain fused to a transmembrane domain and intracellular T-cell costimulatory domains.9 Compared with T-cell receptors, CARs have the advantage of recognizing cancer antigens in an HLA-unrestricted manner. This is of particular relevance for cHL where HLA molecules are downregulated on HRS cells.10 In addition to being able to redirect T cells toward tumor antigens, ex vivo engineering of T cells allows for various modifications to enhance CAR T-cell function and counteract the immunosuppressive tumor microenvironment (TME). These may include coexpression of chemokine receptors on T cells to improve trafficking to tumors, secretion of antitumor cytokines with T cells redirected for universal cytokine killing, and engineering of T cells to be intrinsically resistant to checkpoint inhibition.11
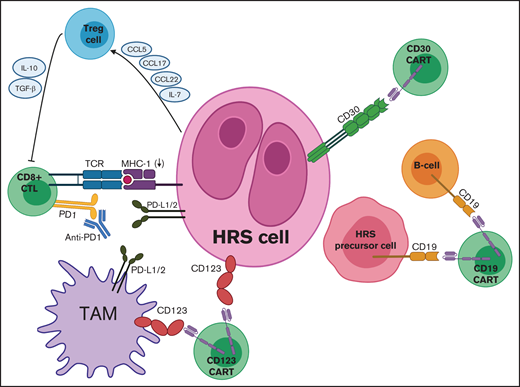
Targets for adoptive T-cell immunotherapies in cHL. Mechanisms of immune evasion depicted include PD-L1 expression by HRS cells and TAMs, downregulation of MHC (also known as HLA), and secretion of various chemokines and cytokines by HRS cells to recruit Treg cells. Adoptive immunotherapy strategies include CD30 CAR T cells, CD19 CAR T cells, and CD123 CAR T cells, which can be combined with PD1 inhibitors to counteract checkpoint blockade. CCL, chemokine (C-C) ligand; TCR, T-cell receptor.
Targets for adoptive T-cell immunotherapies in cHL. Mechanisms of immune evasion depicted include PD-L1 expression by HRS cells and TAMs, downregulation of MHC (also known as HLA), and secretion of various chemokines and cytokines by HRS cells to recruit Treg cells. Adoptive immunotherapy strategies include CD30 CAR T cells, CD19 CAR T cells, and CD123 CAR T cells, which can be combined with PD1 inhibitors to counteract checkpoint blockade. CCL, chemokine (C-C) ligand; TCR, T-cell receptor.
CAR T cells have been successfully used in the treatment of B-cell malignancies, specifically R/R acute lymphoblastic leukemia12 and diffuse large B-cell lymphoma.13 However, the role for CAR T-cell therapy in patients with R/R cHL is still uncertain. Here we review recent reports on adoptive T-cell therapy for cHL with a view toward future development in engineered therapies (Table 1). We focus here on cHL, rather than nodular lymphocyte-predominant Hodgkin lymphoma, because the biology, prognoses, and management for each differs.
Results of adoptive immunotherapy clinical trials in Hodgkin lymphoma
| Therapy | Reference | Phase | Patients, n | Lymphodepletion | Response | Most common toxicities |
|---|---|---|---|---|---|---|
| EBV-specific T cells | Bollard et al38 | 1 | 14 | None | Active disease cohort: ORR 27%; PR 9%; CR 18%; SD 45%. Cohort in remission: 100% disease-free at 10- to 40-mo follow-up | Transient flu-like symptoms (14%) |
| LMP-1/2 specific T cells | Bollard et al37 | 1 | 50 | None | Active disease cohort: ORR 62%; CR 52%; PR 10%; cohort in remission: 2 y EFS 82% | No treatment-related toxicities, although 2 possible inflammatory responses |
| LMP-1/2 specific T cells with DNRII | Bollard et al40 | 1 | 8 | None | Active disease cohort: ORR 43%; PR 14%; SD 57%; patient in remission (n = 1): durable CR up to 2+ years | No treatment-related toxicities |
| Allogeneic LMP-1/2 specific T cells | McLaughlin et al44 | 1 | 26 | None | Active disease: CR 0%; PR 28%; OS of 43% at 2 y; cohort in remission: 2 y EFS 57% at 2 y; 2-y OS 78% | Grade 4 hepatic necrosis in 1/26 (4%) |
| CD30 CAR T cells | Wang et al52 | 1 | 18 | Fludarabine and cyclophosphamide; gemcitabine, mustargen, and cyclophosphamide; or ab-paclitaxel and cyclophosphamide | ORR: 39%; CR: 0%; PR: 39%; SD: 33%. PFS: 6 mo (range: 3-14 mo) | Nausea and vomiting (28%), rash (11%), joint swelling (6%), dizziness (6%), and pneumonitis (6%) |
| CD30 CAR T cells | Ramos et al53 | 1 | 9 | None | ORR: 33%; CR: 33%; SD: 33%; PD: 33%; durable CR up to 2.5+ years | No treatment-related toxicities |
| CD30 CAR T cells | Ramos et al54 | 1/2 | 41 | Bendamustine; bendamustine and fludarabine; or cyclophosphamide and fludarabine | No responses with bendamustine-alone lymphodepletion cohort; responses with fludarabine-based regimen (n = 32): ORR 72%; CR 59%; PR 13%; SD 9%; PD 19% | Rash (48%); grade 1 CRS (24%); grade 3/4 leukopenia (57%), anemia (12%), neutropenia (48%), and thrombocytopenia (26%) |
| CD19 CAR T cells | Svoboda et al58 | Early phase 1 | 4 | Cyclophosphamide | ORR: 50%; CR 25%; PR 25%; SD 25%; PD 25%; at 3 mo, 3/4 had PD and the remainder was taken off trial | Fatigue (75%), headache (75%), confusion (50%); no grade 3 or 4 toxicities |
| Therapy | Reference | Phase | Patients, n | Lymphodepletion | Response | Most common toxicities |
|---|---|---|---|---|---|---|
| EBV-specific T cells | Bollard et al38 | 1 | 14 | None | Active disease cohort: ORR 27%; PR 9%; CR 18%; SD 45%. Cohort in remission: 100% disease-free at 10- to 40-mo follow-up | Transient flu-like symptoms (14%) |
| LMP-1/2 specific T cells | Bollard et al37 | 1 | 50 | None | Active disease cohort: ORR 62%; CR 52%; PR 10%; cohort in remission: 2 y EFS 82% | No treatment-related toxicities, although 2 possible inflammatory responses |
| LMP-1/2 specific T cells with DNRII | Bollard et al40 | 1 | 8 | None | Active disease cohort: ORR 43%; PR 14%; SD 57%; patient in remission (n = 1): durable CR up to 2+ years | No treatment-related toxicities |
| Allogeneic LMP-1/2 specific T cells | McLaughlin et al44 | 1 | 26 | None | Active disease: CR 0%; PR 28%; OS of 43% at 2 y; cohort in remission: 2 y EFS 57% at 2 y; 2-y OS 78% | Grade 4 hepatic necrosis in 1/26 (4%) |
| CD30 CAR T cells | Wang et al52 | 1 | 18 | Fludarabine and cyclophosphamide; gemcitabine, mustargen, and cyclophosphamide; or ab-paclitaxel and cyclophosphamide | ORR: 39%; CR: 0%; PR: 39%; SD: 33%. PFS: 6 mo (range: 3-14 mo) | Nausea and vomiting (28%), rash (11%), joint swelling (6%), dizziness (6%), and pneumonitis (6%) |
| CD30 CAR T cells | Ramos et al53 | 1 | 9 | None | ORR: 33%; CR: 33%; SD: 33%; PD: 33%; durable CR up to 2.5+ years | No treatment-related toxicities |
| CD30 CAR T cells | Ramos et al54 | 1/2 | 41 | Bendamustine; bendamustine and fludarabine; or cyclophosphamide and fludarabine | No responses with bendamustine-alone lymphodepletion cohort; responses with fludarabine-based regimen (n = 32): ORR 72%; CR 59%; PR 13%; SD 9%; PD 19% | Rash (48%); grade 1 CRS (24%); grade 3/4 leukopenia (57%), anemia (12%), neutropenia (48%), and thrombocytopenia (26%) |
| CD19 CAR T cells | Svoboda et al58 | Early phase 1 | 4 | Cyclophosphamide | ORR: 50%; CR 25%; PR 25%; SD 25%; PD 25%; at 3 mo, 3/4 had PD and the remainder was taken off trial | Fatigue (75%), headache (75%), confusion (50%); no grade 3 or 4 toxicities |
Challenge of the immunosuppressive TME in cHL
Knowledge of the biology of cHL and its immunosuppressive TME is essential for developing novel treatment strategies with adoptive T-cell therapy. Malignant HRS cells are intermixed among a much larger population of nonmalignant inflammatory cells, including T and B lymphocytes, eosinophils, mast cells, macrophages, plasma cells, fibroblasts, and other stromal cells.14 HRS cells are derived from post–germinal center B cells and are characterized by the cell surface markers CD15 and CD30, as well as loss of typical B-cell surface markers such as CD19. The HRS cells account for just 1% to 2% of the total tumor cell mass yet thrive in a lymphocyte-rich TME as they have developed various peripheral tolerance strategies to evade the host immune response. One important mechanism of immune evasion in cHL is the interruption of tumor-antigen presentation through downregulation or alteration of HLA class I and II molecules.10,15,16
To maintain their immunologically privileged niche, HRS cells communicate with immune cells in their surrounding TME through the reciprocal secretion of various chemokines and cytokines.17,18 HRS cells produce chemokines such as CCL5, CCL17, and CCL22 and cytokines such as interleukin 7 (IL-7) to recruit T regulatory (Treg) cells, and they express Fas ligand, which can induce apoptosis of tumor-specific cytotoxic T lymphocytes (CTLs).19,20 Treg cells in addition to HRS cells help mediate immune escape through the secretion of IL-10 and transforming growth factor β (TGF-β), which inhibit CTL activity (Figure 1). In addition to Treg cells, TAMs also play a significant role in supporting the immunosuppressive TME. The number of CD68+ and CD163+ TAMs has been shown in some studies to correlate with shortened progression-free survival (PFS) and increased likelihood of relapse after autologous stem cell transplant.21-23
Last, another major mechanism of immune evasion in cHL is the frequent expression of checkpoint ligands by HRS cells and surrounding cells in the tumor microenvironment. HRS cells overexpress programmed death receptor ligand 1 (PD-L1) and PD-L2 through a variety of mechanisms including genomic amplification of chromosome 9p24.1 and Epstein-Barr virus (EBV) infection, which induces latent membrane protein (LMP)-mediated Janus associated kinase-signal transducer and activator of transcription pathway activation.16,24,25 Engagement of PD-L1 and PD-L2 with the PD-1 receptors on T-cell surfaces leads to inhibition of T-cell activation and proliferation. PD-L1 is also frequently expressed in tumor-infiltrating T cells and TAMs, which surround HRS cells to create an immunologically privileged environment.26,27 Cytotoxic T-lymphocyte protein 4 (CTLA4) and lymphocyte activation gene 3 (LAG3) are additional immune checkpoint proteins that negatively regulate the T-cell antitumor response within the TME.28-30
EBV-targeted T cells
The infusion of donor-derived EBV-specific T cells for the treatment of patients with EBV-associated posttransplant lymphoproliferative disease (PTLD) has induced significant clinical responses.31-33 Approximately 40% of patients with cHL have EBV-positive disease.34 Modeling after the successes in PTLD, the use of adoptive immunotherapy in the treatment of cHL was first explored by targeting those with EBV-positive disease. However, various tumor immune evasion strategies need to be overcome to make adoptive immunotherapy an effective therapeutic treatment for those with cHL (Figure 2). In contrast to PTLD, which is seen in immunosuppressed patients and which expresses the entire repertoire of EBV antigens (type III latency pattern), the expression of the EBV-associated antigens on HRS cells follows the type II latency pattern. This pattern is associated with the expression of only the weakly immunogenic EBV antigens LMP 1, LMP 2, EBV nuclear antigen 1 (EBNA 1), and BamH1-A right frame 1 (BARF1).14 The immunodominant EBV antigens, such as EBNA3A, EBNA 3B, and EBNA3C, are downregulated in cHL. Furthermore, EBV-positive HRS cells secrete IL-10, which further inhibits the cytotoxic T-cell response directed at viral antigens.35
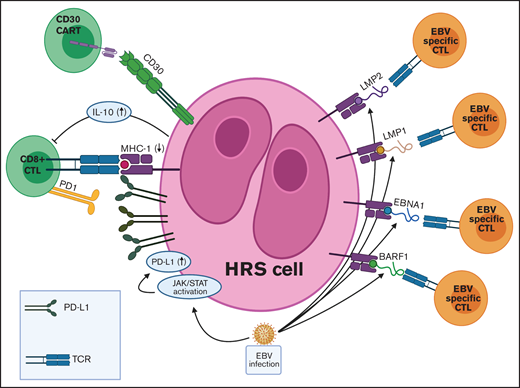
TME in EBV+ cHL and targets for adoptive T-cell immunotherapies. EBV infection is associated with various immune evasion strategies in cHL. (1) EBV infection induces JAK-STAT pathway activation, which leads to increased PD-L1 expression on HRS cells. (2) EBV-infected HRS cells express only the weakly immunogenic EBV antigens (EBNA1, LMP1, LMP2, and BARF1) in a type II latency pattern. (3) EBV+ HRS cells secrete IL-10, which inhibits CTLs directed at viral antigens. Adoptive immunotherapy strategies for EBV+ cHL include autologous and allogeneic EBV-specific CTLs and CAR T cells directed against viral antigens. JAK-STAT, Janus-associated kinase-signal transducer and activator of transcription; TCR, T-cell receptor.
TME in EBV+ cHL and targets for adoptive T-cell immunotherapies. EBV infection is associated with various immune evasion strategies in cHL. (1) EBV infection induces JAK-STAT pathway activation, which leads to increased PD-L1 expression on HRS cells. (2) EBV-infected HRS cells express only the weakly immunogenic EBV antigens (EBNA1, LMP1, LMP2, and BARF1) in a type II latency pattern. (3) EBV+ HRS cells secrete IL-10, which inhibits CTLs directed at viral antigens. Adoptive immunotherapy strategies for EBV+ cHL include autologous and allogeneic EBV-specific CTLs and CAR T cells directed against viral antigens. JAK-STAT, Janus-associated kinase-signal transducer and activator of transcription; TCR, T-cell receptor.
The feasibility of harnessing adoptive immunotherapy to expand EBV-specific CTLs ex vivo for reinfusion was first demonstrated in cHL by Roskrow et al36 in 1998. Subsequent studies of adoptive immunotherapy using EBV-specific CTLs in cHL were performed by Bollard et al.37-39 In their initial study, they generated autologous CTLs directed against a nonspecific array of EBV antigens and infused them into 14 patients with EBV-positive cHL.38 Of 11 patients who had measurable disease at time of CTL infusion, 3 (27%) had no response, 5 (45%) had stable disease (SD), 1 (9%) had a partial response (PR), and 2 (18%) had complete responses (CR). Of the remaining 3 patients with no measurable disease at time of CTL infusion, all remained disease free at 10- to 40-month follow-up. Although this study demonstrated the ability of infused EBV-specific CTLs to expand in vivo, traffic to tumor sites, exhibit antiviral activity, and persist for up to 12 months, the CTLs lacked sufficient antitumor activity as demonstrated by the limited clinical responses. In response, Bollard et al37 refined techniques to improve specificity for the antigens seen in the type II latency pattern of cHL. Although EBNA1 is unable to enter the HLA class I processing pathway, LMP1 and LMP2 antigens provide attractive targets for adoptive immunotherapy with EBV-specific CTLs. Thus, autologous dendritic cells and EBV-transformed B-lymphoblastoid cell lines were transduced with an adenoviral vector expressing either LMP2 or both LMP1 and LMP2. These antigen-presenting cells were then used to expand LMP-specific CTLs, which were infused into 50 patients with type II or III latency EBV-positive lymphomas who were either in remission (29 patients) and were considered high risk for relapse or who had active R/R disease (21 patients).
In the cohort in remission and receiving LMP-specific CTLs as adjuvant therapy, at 8-week follow-up, 27 of 28 patients remained in CR (96%), and 2-year event-free survival (EFS) was 82%. There was a total of 9 deaths, all from nonrelapse causes related to toxicities from prior therapies. In the active disease cohort, 11 of 21 patients achieved CR (52%), and 2 achieved PR (10%). Response rates were similar among patients with cHL compared with those with EBV-positive non-Hodgkin lymphoma (NHL), and the total 2-year EFS rate among patients was approximately 50%. The infusions were well tolerated without any infusional reactions. Patients who achieved response had higher levels of circulating LMP-specific T cells. Moreover, patients who responded showed evidence of epitope spreading, with CTLs developing activity against not just LMP antigens, but non-viral tumor antigens as well. Of the total cohort in this study, 25 patients had cHL, 8 with active disease, and 17 in a first or later remission. Notably, although cHL is associated with type II latency pattern of EBV antigen expression, 5 of the 25 HL patients had type III latency because of comorbid immunodeficiency. Of the 8 with active disease (5 with type II latency and 3 with type III latency), the overall response rate (ORR) was 50% (3 CR, 1 PR). All 3 patients with type III latency pattern achieved CR, whereas the ORR for those with type II latency was just 10%, suggesting potential differences in tumor immune evasion that are more easily overcome in patients that are immunosuppressed.
To further improve the efficacy of EBV-specific CTL therapy, Bollard et al40 sought to target the immunosuppressive TME seen in cHL by transducing EBV-specific CTLs with a dominant-negative TGF-β type 2 receptor (DNRII). TGF-β is a cytokine secreted by cells in the TME that inhibits cytotoxic T-cell activity against tumors by suppressing T-cell proliferation, activation, and effector functions. Abrogation of TGF-β activity in various preclinical studies has been shown to improve the efficacy of adoptive immunotherapy in various preclinical models.41-43 In this phase 1 study, LMP-specific CTLs transduced with DNRII were infused into 8 patients with extensively pretreated R/R EBV-positive cHL. Of the 7 with active disease, ORR was 43%: 2 (29%) achieved CR, 1 achieved PR (14%), and 4 had SD (57%). The 1 patient who was in remission at time of infusion but was high risk for relapse remained in complete remission for greater than 2 years. Two of the patients treated had been previously treated with LMP-specific CTLs that were not transduced with DNRII39 and showed greater responses to infusion with DNRII CTLs. Although comparisons are limited by the small number of patients, the higher ORR in this cohort of more extensively treated and immunocompetent patients with cHL is encouraging and further emphasizes the importance of a therapeutic approach targeting both malignant cells and the TME.
In addition to autologous T-cell adoptive immunotherapy, infusion of donor-derived allogeneic EBV-specific cytotoxic T cells in patients with EBV-positive lymphoma has also been shown in small trials to be safe and produce clinical responses.44,45 In 1 study, 26 patients with EBV-positive cHL, NHL, or natural killer/T-cell lymphoproliferative disease received allogeneic LMP-specific T-cell infusions either for active disease (7 patients) or as adjuvant therapy (19 patients) after allogeneic stem cell transplantation. Of the entire cohort, there were 6 patients with cHL, 5 of which were in remission at the time of infusion and 1 who was treated for active disease. Of the 5 patients with cHL treated with allogeneic LMP-specific T cells as adjuvant therapy, 3 were still alive and in remission at 3-year follow up. The patient treated for active disease had stable disease 8 weeks after T-cell infusion and was alive with disease at 3-year follow-up.
With the clinical responses seen with the use of adoptive T-cell immunotherapy in patients with EBV-positive cHL, there are multiple current clinical trials evaluating novel strategies (Table 2). Given that the manufacturing process for EBV-specific CTLs is prohibitive for those with rapidly progressive disease needing more urgent treatment, the use of partially matched banked EBV-specific CTLs is currently being explored in a clinical trial (NCT02287311). The CTLs used in this study are specific for LMP, BARF-1, and EBNA-1. Although the immunogenicity of the EBV antigens EBNA-1 and LMP has been more readily studied, BARF-1 has only more recently been recognized as an additional immunotherapy target for patients with EBV type II and III latency malignancies.46 LMP-specific CAR T cells have been developed in preclinical in vivo models of EBV-positive nasopharyngeal carcinoma but have yet to be tried in the clinical setting.47 Last, the success of adoptive immunotherapy in patients with EBV-positive malignancies has also led to a trial of adoptive CTLs targeted against tumor-associated antigens (NY-ESO-1, MAGEA4, PRAME, Survivin, and SSX) in patients with R/R cHL, regardless of EBV status (NCT01333046).
Ongoing clinical trials of adoptive immunotherapy in Hodgkin lymphoma
| Treatment | Trial identifier | Trial site | Trial phase | Dose/conditioning | Study summary |
|---|---|---|---|---|---|
| CD30 CAR T cells | NCT03049449 | National Cancer Institute (NCI) | 1 | Dose: 0·3-1 × 106 cells/kg; conditioning: cyclophosphamide and fludarabine | Evaluate the safety and feasibility of anti-CD30 CARTs in patients with advanced CD30-expressing lymphomas |
| CD30 CAR T cells | ChiCTR2000041436 | Cancer Hospital of Guangxi Medical University, China | 1 | Not available | Evaluate the safety and efficacy of CAR T-cell therapy targeting CD19,CD20,CD22,CD30,CD79B,CD99,CD38,CD7, or BCMA for relapsed/refractory tumors of hematopoietic and lymphoid tissues |
| CD30 CAR T cells | NCT02690545 | UNC Lineberger Comprehensive Cancer Center | 1b/2 | Dose: 1-2 × 108 cells/m2; conditioning: bendamustine and fludarabine | Evaluate the safety and efficacy of CD30 CAR T-cells in patients with CD30+ R/R HL and NHL |
| CD30 CAR T cells | NCT02917083 | Baylor College of Medicine | 1 | Dose: 0·2-2 × 108 cells/m2; conditioning: cyclophosphamide and fludarabine | Evaluate the safety and efficacy of CD30 CAR T-cells in patients with CD30+ R/R HL and NHL |
| CD30.CCR4 CAR T cells | NCT03602157 | UNC Lineberger Comprehensive Cancer Center | 1 | Dose: 0·2-2 108 cells/m2; conditioning: fludarabine and bendamustine | Evaluate the safety and tolerability of CD30.CCR4 CAR T-cells +/− CD30 CAR T-cells in patients with R/R CD30+ HL or CTCL |
| CD30 and CD19 CAR T cells | ChiCTR2000028922 | The Third Affiliated Hospital of Kunming Medical University, China | Early phase 1 | Not available | Evaluate the feasibility and efficacy of combined use of CD19 and CD30 CAR T-cells in patients with R/R HL |
| CD30 CAR T cells | NCT02958410 | Southwest Hospital, China | 1/2 | Not available | Evaluate the safety and efficacy of CD30 CAR T cells in patients with R/R CD30+ lymphomas |
| CD30 CAR T cells | NCT04008394 | Wuhan Union Hospital, China | 1 | Not available | Evaluate the safety and efficacy of CD30 CAR T cells in patients with R/R CD30+ lymphomas |
| CD30 CAR T cells | ChiCTR-OPN-16009069 | Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, China | 1 | Not available | Evaluate the safety and efficacy of CD30 CAR T cells in patients with R/R CD30+ lymphomas |
| CD30 CAR T cells | NCT02663297 | UNC Lineberger Comprehensive Cancer Center | 1 | Dose: 0·2-2 × 108 cells/m2 | Evaluate the safety and tolerability of CD30 CAR T-cells for prevention of relapse after autologous stem cell transplant in patients with CD30+ lymphomas |
| CD30 CAR T cells | NCT04653649 | l'Hospital de la Santa Creu i Sant Pau, Spain | 1/2a | Dose: 3-10 × 106/kg | Evaluate the safety, maximum-tolerated dose, and response rate of CD30 CAR T-cells in patients with R/R CD30+ HL or NHL |
| EBV CTLs expressing CD30 CARs | NCT01192464 | Baylor College of Medicine | 1 | Dose: 0·2-1 × 108 cells/m2 | Evaluate the safety and efficacy of autologous EBV-specific cytotoxic T-lymphocytes genetically modified to express a CD30 CAR in patients with R/R HL or NHL |
| Allogeneic CD30 CAR EBV-specific T lymphocytes | NCT04288726 | Baylor College of Medicine | 1 | Dose: 0·4-4 × 108 cells/m2 | Evaluate the dose-limiting toxicity rate and response to allogeneic CD30 CARs engineered onto EBV-specific T cells in patients with CD30+ HL, NHL, ALCL, or peripheral T-cell lymphoma |
| CD30 CAR T cells | ChiCTR2000030843 | Beijing Boren Hospital, China | Early phase 1 | Not available | Evaluate the safety and efficacy of CD30 CAR T cells in patients with R/R CD30+ lymphomas |
| CD30 CAR T cells | NCT02259556 | Chinese PLA General Hospital, China | Phase 1/2 | Dose: not available; conditioning: cyclophosphamide and fludarabine | Evaluate the safety and efficacy of CD30 CAR T cells in patients with R/R CD30+ lymphomas |
| CD30 CAR T cells | NCT02274584 | Peking University Cancer Hospital, China and University of Florida | 1/2 | Not available | Evaluate the safety and efficacy of CD30 CAR T-cells engineered with a self-withdrawal mechanism (FKBP-iCasp9) in patients with R/R CD30+ lymphomas |
| CD30 CAR T cells | NCT04268706 | Tessa Therapeutics | 2 | Conditioning: fludarabine and bendamustine | Evaluate the safety and efficacy of CD30 CAR T-cells in patients with R/R CD30+ HL |
| LMP 1/2 CTLs | NCT01956084 | Children’s National Medical Center | 1 | Dose: 1-5 × 107 cells/m2 | Evaluate the dose-limiting toxicities and survival of LMP-specific CTLs in patients with EBV+HL or NHL after allogeneic stem cell transplant |
| PD-1 knockout EBV-CTLs | NCT03044743 | The Comprehensive Cancer Center of Nanjing Drum Tower Hospital, China | 1/2 | Dose: 2 × 107 cells/kg; conditioning: fludarabine and cyclophosphamide | Evaluate the safety of EBV-CTLs that have been knocked out for PD1 by the CRISP-Cas9 system, in treating patients with EBV+ advanced malignancies |
| EBV CTLs | NCT01555892 | Baylor College of Medicine | 1 | Dose: 1 × 108 cells/m2 | Evaluate the toxicity of escalating doses and anti-viral/anti-tumor effects of autologous LMP, BARF1, and EBNA1-specific T-lymphocytes in patients with EBV-associated HL |
| Tumor-associated antigen-specific CTLs | NCT01333046 | Baylor College of Medicine | 1 | Dose: 0.5-2 × 107 cells/m2 | Evaluate the safety and expansion, persistence, and anti-tumor effects of adoptively-transferred tumor-associated antigen (PRAME, SSX, MAGE, NY-ESSO, Survivin) -specific CTLs +/− azacytidine in patients with R/R HL or NHL |
| LMP, BARF1, and EBNA1-specific CTLs | NCT02287311 | Baylor College of Medicine | 1 | Dose: 0.4-1.5 × 108 cells/m2; conditioning: cyclophosphamide and fludarabine if circulating T cells are high | Evaluate the safety and dose-limiting toxicity of banked allogeneic, partially HLA-matched rapid EBV-specific T cells in patients with R/R EBV+ HL or NHL |
| Treatment | Trial identifier | Trial site | Trial phase | Dose/conditioning | Study summary |
|---|---|---|---|---|---|
| CD30 CAR T cells | NCT03049449 | National Cancer Institute (NCI) | 1 | Dose: 0·3-1 × 106 cells/kg; conditioning: cyclophosphamide and fludarabine | Evaluate the safety and feasibility of anti-CD30 CARTs in patients with advanced CD30-expressing lymphomas |
| CD30 CAR T cells | ChiCTR2000041436 | Cancer Hospital of Guangxi Medical University, China | 1 | Not available | Evaluate the safety and efficacy of CAR T-cell therapy targeting CD19,CD20,CD22,CD30,CD79B,CD99,CD38,CD7, or BCMA for relapsed/refractory tumors of hematopoietic and lymphoid tissues |
| CD30 CAR T cells | NCT02690545 | UNC Lineberger Comprehensive Cancer Center | 1b/2 | Dose: 1-2 × 108 cells/m2; conditioning: bendamustine and fludarabine | Evaluate the safety and efficacy of CD30 CAR T-cells in patients with CD30+ R/R HL and NHL |
| CD30 CAR T cells | NCT02917083 | Baylor College of Medicine | 1 | Dose: 0·2-2 × 108 cells/m2; conditioning: cyclophosphamide and fludarabine | Evaluate the safety and efficacy of CD30 CAR T-cells in patients with CD30+ R/R HL and NHL |
| CD30.CCR4 CAR T cells | NCT03602157 | UNC Lineberger Comprehensive Cancer Center | 1 | Dose: 0·2-2 108 cells/m2; conditioning: fludarabine and bendamustine | Evaluate the safety and tolerability of CD30.CCR4 CAR T-cells +/− CD30 CAR T-cells in patients with R/R CD30+ HL or CTCL |
| CD30 and CD19 CAR T cells | ChiCTR2000028922 | The Third Affiliated Hospital of Kunming Medical University, China | Early phase 1 | Not available | Evaluate the feasibility and efficacy of combined use of CD19 and CD30 CAR T-cells in patients with R/R HL |
| CD30 CAR T cells | NCT02958410 | Southwest Hospital, China | 1/2 | Not available | Evaluate the safety and efficacy of CD30 CAR T cells in patients with R/R CD30+ lymphomas |
| CD30 CAR T cells | NCT04008394 | Wuhan Union Hospital, China | 1 | Not available | Evaluate the safety and efficacy of CD30 CAR T cells in patients with R/R CD30+ lymphomas |
| CD30 CAR T cells | ChiCTR-OPN-16009069 | Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, China | 1 | Not available | Evaluate the safety and efficacy of CD30 CAR T cells in patients with R/R CD30+ lymphomas |
| CD30 CAR T cells | NCT02663297 | UNC Lineberger Comprehensive Cancer Center | 1 | Dose: 0·2-2 × 108 cells/m2 | Evaluate the safety and tolerability of CD30 CAR T-cells for prevention of relapse after autologous stem cell transplant in patients with CD30+ lymphomas |
| CD30 CAR T cells | NCT04653649 | l'Hospital de la Santa Creu i Sant Pau, Spain | 1/2a | Dose: 3-10 × 106/kg | Evaluate the safety, maximum-tolerated dose, and response rate of CD30 CAR T-cells in patients with R/R CD30+ HL or NHL |
| EBV CTLs expressing CD30 CARs | NCT01192464 | Baylor College of Medicine | 1 | Dose: 0·2-1 × 108 cells/m2 | Evaluate the safety and efficacy of autologous EBV-specific cytotoxic T-lymphocytes genetically modified to express a CD30 CAR in patients with R/R HL or NHL |
| Allogeneic CD30 CAR EBV-specific T lymphocytes | NCT04288726 | Baylor College of Medicine | 1 | Dose: 0·4-4 × 108 cells/m2 | Evaluate the dose-limiting toxicity rate and response to allogeneic CD30 CARs engineered onto EBV-specific T cells in patients with CD30+ HL, NHL, ALCL, or peripheral T-cell lymphoma |
| CD30 CAR T cells | ChiCTR2000030843 | Beijing Boren Hospital, China | Early phase 1 | Not available | Evaluate the safety and efficacy of CD30 CAR T cells in patients with R/R CD30+ lymphomas |
| CD30 CAR T cells | NCT02259556 | Chinese PLA General Hospital, China | Phase 1/2 | Dose: not available; conditioning: cyclophosphamide and fludarabine | Evaluate the safety and efficacy of CD30 CAR T cells in patients with R/R CD30+ lymphomas |
| CD30 CAR T cells | NCT02274584 | Peking University Cancer Hospital, China and University of Florida | 1/2 | Not available | Evaluate the safety and efficacy of CD30 CAR T-cells engineered with a self-withdrawal mechanism (FKBP-iCasp9) in patients with R/R CD30+ lymphomas |
| CD30 CAR T cells | NCT04268706 | Tessa Therapeutics | 2 | Conditioning: fludarabine and bendamustine | Evaluate the safety and efficacy of CD30 CAR T-cells in patients with R/R CD30+ HL |
| LMP 1/2 CTLs | NCT01956084 | Children’s National Medical Center | 1 | Dose: 1-5 × 107 cells/m2 | Evaluate the dose-limiting toxicities and survival of LMP-specific CTLs in patients with EBV+HL or NHL after allogeneic stem cell transplant |
| PD-1 knockout EBV-CTLs | NCT03044743 | The Comprehensive Cancer Center of Nanjing Drum Tower Hospital, China | 1/2 | Dose: 2 × 107 cells/kg; conditioning: fludarabine and cyclophosphamide | Evaluate the safety of EBV-CTLs that have been knocked out for PD1 by the CRISP-Cas9 system, in treating patients with EBV+ advanced malignancies |
| EBV CTLs | NCT01555892 | Baylor College of Medicine | 1 | Dose: 1 × 108 cells/m2 | Evaluate the toxicity of escalating doses and anti-viral/anti-tumor effects of autologous LMP, BARF1, and EBNA1-specific T-lymphocytes in patients with EBV-associated HL |
| Tumor-associated antigen-specific CTLs | NCT01333046 | Baylor College of Medicine | 1 | Dose: 0.5-2 × 107 cells/m2 | Evaluate the safety and expansion, persistence, and anti-tumor effects of adoptively-transferred tumor-associated antigen (PRAME, SSX, MAGE, NY-ESSO, Survivin) -specific CTLs +/− azacytidine in patients with R/R HL or NHL |
| LMP, BARF1, and EBNA1-specific CTLs | NCT02287311 | Baylor College of Medicine | 1 | Dose: 0.4-1.5 × 108 cells/m2; conditioning: cyclophosphamide and fludarabine if circulating T cells are high | Evaluate the safety and dose-limiting toxicity of banked allogeneic, partially HLA-matched rapid EBV-specific T cells in patients with R/R EBV+ HL or NHL |
CD30 CAR T cells
CD30 is a member of the tumor necrosis factor receptor superfamily that, when stimulated, results in downstream activation of the nuclear factor-κB and MAPK pathways that have antiapoptotic and prosurvival effects on cells.48 CD30 is highly expressed on HRS and anaplastic large-cell lymphoma (ALCL) cells yet only minimally expressed on nonmalignant activated B and T cells, thus limiting on-target off-tumor toxicity and making it an ideal therapeutic target. The success of BV, a CD30 antibody conjugated to the cytotoxic antimicrotubule agent monomethyl auristatin E, in patients with cHL has also further propelled the interest in CD30-targeting CAR T cells.49-51
There are currently 3 published studies of CD30-targeting CAR T cells. A phase 1 trial conducted by Wang et al52 enrolled and infused 18 patients with heavily pretreated CD30+ R/R lymphoma (17 with cHL, 1 with ALCL). All patients had progressive disease (PD) at the time of CAR T-cell infusion, and 5 (28%) had previously received BV. The patients received 1 of 3 lymphodepleting (LD) regimens (fludarabine and cyclophosphamide, gemcitabine and mustargen and cyclophosphamide, or ab-paclitaxel and cyclophosphamide) followed by infusion of a mean of 1·56 × 107 CAR T cells/kg. The treatment was well tolerated, with only 2 patients experiencing grade 3 or 4 toxicities (transaminitis and left ventricular systolic dysfunction), which were attributed to prior chemotherapy. The most common adverse events were nausea and vomiting (28%), rash (11%), joint swelling (6%), dizziness (6%), and pneumonitis (6%). Of the 18 patients enrolled, 7 (39%) achieved PR, 6 (33%) had SD, and 5 (27%) had PD. The median PFS was 6 months (range, 3-14 months). Five patients received a second CAR T-cell infusion. Of these, 3 maintained PR, 1 maintained SD, and 1 had PR after having SD after the first infusion.
Another report was of a phase 1 dose-escalation study assessing the safety of anti-CD30 CAR T cells without LD chemotherapy in 9 patients with CD30+ lymphoma (6 with cHL, 1 with diffuse large B cell lymphoma evolved to cHL, and 2 with ALCL).53 Patients were infused with 1 of 3 dose levels, from 0·2 × 108 to 2 × 108 CD30 CAR T cells/m2. All 9 patients were heavily pretreated, and 7 had previously received BV. Treatment was well tolerated at all dose levels, with an ORR of 33%. Of the 9 patients, 3 had clinical responses (2 CR, 1 continued CR), 3 had SD, and 3 had PD.
A recently published report of 2 parallel phase 1/2 trials at the University of North Carolina (UNC) and Baylor College of Medicine investigated anti-CD30 CAR T cells in 41 patients with R/R cHL.54 Patients enrolled had a median of 7 prior lines of therapy; most received prior BV (90%) and/or a checkpoint inhibitor (81%). Patients received LD 2 to 5 days before CAR T-cell infusion with either bendamustine, bendamustine and fludarabine, or cyclophosphamide and fludarabine. Patients received 1 of 3 dose levels (2 × 107 CAR T cells/m2, 1 × 108 CAR T cells/m2, or 2 × 108 CAR T cells/m2). There were no dose-limiting toxicities, and cytokine release syndrome (CRS) occurred in 24% of patients, although all were grade 1 and resolved spontaneously. A transient rash occurred in 48% of patients, mostly in the group that got cyclophosphamide and fludarabine lymphodepletion (82%). Other notable grade 3/4 toxicities were mostly hematologic. Thrombocytopenia was significant, with grade 3/4 thrombocytopenia not resolving by day 28 in 10 (24%) patients, 4 (10%) of which had not resolved by 3 months. Of the patients with grade 3/4 neutropenia, 4 (10%) had not resolved by day 28, although all resolved by 3 months. One common concern raised with CAR T-cell therapy is that CD30 CAR T cells could impair cell-mediated immunity given that CD30 is in a small fraction of activated B and T cells. However, in all 3 reported studies, there was no significant increase in infection, particularly viral infections.
Among the 32 patients who underwent LD with a fludarabine-based regimen and were evaluable for disease response, ORR was 72%; 19 (59%) achieved CR, 4 (13%) achieved PR, 3 (9%) had SD, and 6 (19%) had PD at the time of first response assessment. The 1-year PFS for those with active disease at the time of treatment was 36% and significantly longer in those receiving a fludarabine-based lymphodepleting regimen vs bendamustine alone (P = .0002). CAR T-cell persistence was higher in patients who received the 2 × 108 CAR T cells/m2 dose (P < .001), irrespective of LD regimen. Although increased CAR T-cell dosing correlated with peak expansion, there was no significant correlation with clinical response.
Given the promise seen with the initial trials of CD30 CAR T cells, there are currently multiple clinical trials of CD30 CAR T cells in patients with R/R cHL (Table 2). One novel strategy that is being tested in a clinical trial (NCT03602157) is the use of CD30 CAR T cells engineered to coexpress CCR4. HRS cells surround themselves with an immune privileged niche by producing CCL17 and CCL22, which bind to their ligand CCR4 on Treg cells.55-57 CD30 CAR T cells engineered to coexpress an anti-CD30 CAR and CCR4 have been shown in a mouse model of cHL to improve tumor trafficking and antitumor activity compared with CD30 CAR T cells alone.57 Additionally, the use of an anti-CCR4 monoclonal antibody was effective in depleting CCR4+ T cells and preventing migration of Treg cells in vitro, suggesting a possible role for combination therapy with an anti-CCR4 monoclonal antibody and CAR T cells. Last, there are currently 2 active clinical trials investigating the use of EBV-specific CTLs engineered to express an anti-CD30 CAR. In 1 trial (NCT01192464), autologous CD30 CAR EBV-specific CTLs are given to patients with both CD30+ cHL and EBV seropositivity. In another (NCT04288726), allogeneic CD30 CAR EBV-specific CTLs are given to patients with CD30+ cHL, regardless of their EBV status.
Approaches that target the tumor microenvironment
Given that cellular therapy strategies that directly target HRS cells have had variable success and responses may be blunted by immune escape mechanisms of the surrounding inflammatory milieu, strategies that target cells in the immunosuppressive TME warrant further exploration. Although HRS cells do not express CD19, CAR T cells that target CD19 (CART19) have been piloted in patients with R/R cHL with the goal of targeting nonmalignant B cells in the TME and circulating CD19+ B cells in the blood that may represent precursor HRS cells.58,59 In a pilot trial using this strategy, 4 patients with R/R cHL who had failed at least 1 salvage therapy and had no curative therapeutic options received LD with cyclophosphamide followed by 6 infusions of nonviral RNA anti-CD19 CAR T cells.58 The therapy was well tolerated with no grade 3 or 4 nonhematologic adverse events and no CRS. Of the 4 patients, at 1-month follow-up, 1 had CR, 1 had PR, 1 had SD, and 1 had PD, but the responses seen were transient.
CAR T cells against CD123 have also been considered in cHL. CD123 is the α chain of the IL-3 receptor and is present on ∼60% of cHL cell populations.60 IL-3 serves as a growth and antiapoptotic factor for HRS cells.61 Moreover, CD123 is present on other cells in the supportive TME, particularly the M2-type TAMs.21,60 In contrast to the M1-type macrophages, which work as anti-infection and antitumor macrophages, the M2-type macrophages suppress the immune response and promote tumor proliferation. Ruella et al62 developed an anti-CD123 CAR T cell to simultaneously target HRS cells and disrupt the TME. In this preclinical study, the authors demonstrated that anti-CD123 CAR T cells were able to eradicate cHL cells both in vitro and in 2 tumor xenografts of disseminated cHL, develop immunologic memory, and resist suppression by M2 macrophages.
One potential strategy for future studies would be to look at the combined infusion of CD30 CAR T cells with CD19 or CD123 CAR T cells. This combination strategy offers the opportunity to not only dually target antigens but also to counter any potential antigen escape. However, in the study of CD30 CAR T cells by Ramos et al,54 CD30 expression persisted in patients with relapsed disease, suggesting that antigen escape is unlikely the etiology of disease recurrence.63 The strategy of combination CAR T cells targeting multiple antigens is being explored in many studies, including one targeting CD19/CD30 and CD19/CD123 in patients with refractory B-cell malignancies (NCT03125577).
Conclusions and future perspectives
The treatment landscape for patients with R/R cHL has rapidly changed over the last decade with advancements in immunotherapy led by improved understanding of the PD-1/PDL-1 axis and the complex interplay between HRS cells and the surrounding immunosuppressive TME. Driven by the success of CAR T-cell therapy in other hematologic malignancies, there is a burgeoning interest in whether CAR T-cell therapy can reliably find its place in the treatment of R/R cHL. The results of clinical trials with EBV-specific CTLs and anti-CD30 CAR T cells to target tumor-associated antigens in patients with extensively pretreated and multiply relapsed cHL are thus far encouraging and highlight the potential for CAR T-cell therapy in cHL (Table 1).
To build on these successes, therapeutic strategies that simultaneously target HRS cells and disrupt the immunosuppressive cross talk between HRS cells and the TME should be considered to enhance the antitumor properties and improve persistence of adoptively transferred T cells. Various preclinical studies have demonstrated enhanced activity of CAR T cells that have been combined with checkpoint inhibitors,64 engineered to secrete anti–PD-1 checkpoint inhibitors,65 or genetically altered with the clustered regularly interspaced short palindromic repeats associated with Cas9 endonuclease (CRISPR-Cas9) gene-editing system to disrupt PD-1 expression.66,67 These techniques can be applied to the use of CAR T-cell therapy in cHL, where in 1 study, approximately 33% of CD30 CAR T cells demonstrated PD-L1 expression.53 Currently, there is an ongoing clinical trial (NCT03044743) evaluating the safety and efficacy of PD-1 knockout EBV-specific CTLs in treating EBV-positive cHL. There is also a clinical trial (NCT04134325) assessing the role of anti–PD-1 therapy in patients who have previously progressed on anti–PD-1 therapy and also progressed after receiving anti-CD30 CAR T cells. Finally, as modeled by the studies of anti-CD19 and anti-CD123 CAR T cells, additional approaches that target not just HRS cells but also the supportive cells in the TME or precursor tumor cells should be explored as well.58,62 With these additional strategies, we may see more significant advancements in the use of CAR T-cell therapy for patients with R/R cHL.
Authorship
Contribution: C.H. contributed to the literature review and writing the original draft of the manuscript; J.S. contributed to the design of the review, literature review, and review and editing of the manuscript; M.R. and B.L.L. contributed to review and editing of the manuscript; and J.S., M.R., and B.L.L. reviewed the manuscript for data and clinical accuracy.
Conflict-of-interest disclosure: M.R. has grants/contracts with Abclon, Beckman-Coulter, and Nanostring; receives consulting fees from Bristol-Myers Squibb and Nanostring; has patents planned, issued, or pending with CART Technologies, University of Pennsylvania, partly licensed to Novartis and Tmunity; and serves on the data safety monitoring board or advisory board of Abclon and Bayer. J.S. has grants/contracts with BMS, Incyte, Merck, Novartis, Astra-Zeneca, Adaptive, Pharmacyclics, TG, and Seattle Genetics and serves on the data safety monitoring board or advisory board for Seattle Genetics, Imbrium, Genmab, BMS, Atara, Adaptive, and Astra-Zeneca. B.L.L. reports personal fees from Avectas, personal fees from Patheon/ThermoFisher Viral Vector Services, personal fees from In8bio, personal fees from Vycellix, other from Tmunity Therapeutics, personal fees from Immuneel, personal fees from Ori Biotech, personal fees from Akron Biotech, personal fees from Immusoft, and personal fees from TerumoBCT, outside the submitted work; In addition, B.L.L. has a patent “Methods for treatment of cancer” (US 8906682; US 8916381; US 9101584) with royalties paid to University of Pennsylvania, a patent “Compositions for treatment of cancer” (US 8911993; US 9102761; US 9102760) with royalties paid to University of Pennsylvania, a patent “Method for treating chronic lymphocytic leukemia (CC)” (US 9161971) with royalties paid to University of Pennsylvania, a patent “Compositions and methods for treatment of cancer” (US 9464140; US 9518123; US 9481728; US 9540445) with royalties paid to University of Pennsylvania, a patent “Use of chimeric antigen receptor-modified T cells to treat cancer” (US 9328156; US 9499629) with royalties paid to University of Pennsylvania, and a patent “Methods for assessing the suitability of transduced T cells for administration” (US 9572836) with royalties paid to University of Pennsylvania. C.H. declares no competing financial interests.
Correspondence: Jakub Svoboda, Perelman Center for Advanced Medicine, 34th & Civic Center Blvd, 12th Floor South Pavilion, Philadelphia, PA 19104; e-mail: jakub.svoboda@pennmedicine.upenn.edu.

