

Key Points
Engineered IKZF1 perturbations result in a stem cell–like expression signature, enhanced engraftment in vivo, and multidrug resistance.
SAMHD1 is downregulated after IKZF1 knockout resulting in enhanced sensitivity to cytarabine, which is rescuable via SAMHD1 restoration.

Abstract
IKAROS family zinc finger 1 (IKZF1) alterations represent a diverse group of genetic lesions that are associated with an increased risk of relapse in B-cell acute lymphoblastic leukemia. Due to the heterogeneity of concomitant lesions, it remains unclear how IKZF1 abnormalities directly affect cell function and therapy resistance, and whether their consideration as a prognostic indicator is valuable in improving outcome. CRISPR/Cas9 strategies were used to engineer multiple panels of isogeneic lymphoid leukemia cell lines with a spectrum of IKZF1 lesions to measure changes in chemosensitivity, gene expression, cell cycle, and in vivo engraftment that can be linked to loss of IKAROS protein. IKZF1 knockout and heterozygous null cells displayed relative resistance to a number of common therapies for B-cell acute lymphoblastic leukemia, including dexamethasone, asparaginase, and daunorubicin. Transcription profiling revealed a stem/myeloid cell–like phenotype and JAK/STAT upregulation after IKAROS loss. A CRISPR homology-directed repair strategy was also used to knock-in the dominant-negative IK6 isoform into the endogenous locus, and a similar drug resistance profile, with the exception of retained dexamethasone sensitivity, was observed. Interestingly, IKZF1 knockout and IK6 knock-in cells both have significantly increased sensitivity to cytarabine, likely owing to marked downregulation of SAMHD1 after IKZF1 knockout. Both types of IKZF1 lesions decreased the survival time of xenograft mice, with higher numbers of circulating blasts and increased organ infiltration. Given these findings, exact specification of IKZF1 status in patients may be a beneficial addition to risk stratification and could inform therapy.
Introduction
Approximately 20% of pediatric patients and the majority of adults with B-cell acute lymphoblastic leukemia (B-ALL) experience relapse, and prognosis after relapse is very poor.1,2 Identifying those at risk for treatment failure and devising novel approaches to mitigate their relapse risk is imperative to improving outcomes.
In B-ALL, deletions and mutations of the IKAROS family zinc finger 1 (IKZF1) gene are associated with an increased risk of relapse.3 IKZF1 encodes the protein IKAROS, which is a master lymphoid regulatory transcription factor4,5 and chromatin remodeler.6 One of the earliest regulators of lymphoid lineage identity,7 IKAROS is required for terminal B-cell maturation,8,9 and its loss of function is an important factor in differentiation-arrested, immature B-cell leukemias.
Various types of lesions in IKZF1 may occur in patients with B-ALL, including monoallelic or biallelic deletion of the entire gene, intragenic deletions, and loss-of-function mutations.10 The most common intragenic deletion (found in ∼23% of B-ALL cases) results in exclusive expression of the short IK6 isoform,11,12 secondary to deletion of a 50 kb intragenic region13 (containing exons 4-7), and the resulting loss of 4 critical DNA-binding zinc fingers.14 IK6 retains the capacity to dimerize with residual wild-type (WT) IKAROS, inhibiting its function; thus, IK6 is a dominant-negative inhibitor of IKAROS function. When virally overexpressed, IK6 results in widespread transcriptional changes that promote proliferation, as cells are able to bypass typical metabolic restrictions imposed by tumor suppressive transcriptional regulators.15,16 However, IK6 alone is insufficient to induce leukemia in normal human hematopoietic cells, and it asserts different regulatory functions.17 In addition, mouse studies using inducible deletion of only 2 of these DNA-binding zinc fingers (exon 5) have shown that expression of this isoform with limited DNA-binding ability can promote carcinogenesis by mediating the transition to stromal independence via disruption of integrin signaling pathways.18
Although IKZF1 mutations and deletions are clinically correlated with poor outcome and increased risk of relapse, indicating that IKZF1 status could be a useful prognostic marker, clinical trials to modulate therapeutic intensity based on IKZF1 status have produced mixed results.19–21 How the broad spectrum of IKZF1 genetic variants seen in B-ALL affect chemosensitivity and outcome and whether IKZF1 status is a relevant prognostic criterion only in the context of certain driver translocations/mutations remain unclear.22
Some previously published in vitro studies using RNA interference (RNAi)-mediated knockdown of IKZF1 have shown evidence of resistance to corticosteroids,11,23 but others report no change in chemosensitivity.24 These mixed results may be in part due to variable reduction in IKAROS protein with RNAi strategies. In addition, previous studies on the impact of the IK6 isoform in B-ALL have been limited by reliance on viral overexpression14,15,25,26 that may not accurately model the relative expression level of IK6 to other IKAROS isoforms. To overcome the limitations of these previous model systems, CRISPR/Cas9 strategies were used to generate a series of human B-ALL cell lines with IKZF1 lesions mirroring those found in patients. We generated isogenic cell lines heterozygous or null for IKZF1 by targeting early exons with efficient single guide RNAs (sgRNA). A CRISPR/Cas9 homology-directed repair (HDR) strategy was also developed in which the IK6 isoform is knocked-in to the endogenous locus. This system allows for the expression of IK6 under the complete control of the endogenous promoter while simultaneously removing the expression of one WT allele, genetically recapitulating the heterozygous IK6-generating intragenic deletion occurring in patients with B-ALL. With this series of human B-ALL cell lines, we investigated the genotype-specific differences in multidrug resistance, gene expression profiles, in vivo engraftment, and cell cycle kinetics.
Methods
Generation of isogeneic human B-ALL cell lines with IKZF1 lesions
Using CRISPRscan (http://www.crisprscan.org),27 we selected a series of 6 sgRNAs targeting exon 2 (the first coding exon) or exon 3 of IKZF1. Only sgRNAs were chosen with no predicted off-target sites and with maximum cleavage efficiency; they were synthesized by in vitro transcription as previously described28 using the HiScribe T7 High Yield RNA Synthesis Kit (New England Biolabs, E2040S) or, alternatively, were commercially synthesized with 2′-O-methyl 3'phosphorothioate modifications in the first and last 3 nucleotides (Synthego). Recombinant high-fidelity Cas9 protein (Integrated DNA Technologies, #1081061) was selected to further minimize the incidence of off-target cleavage.
We introduced the CRISPR/Cas9 system by electroporation with sgRNA-Cas9 ribonucleoprotein complexes (RNPs) to generate IKZF1 mutant clones as previously described28 using the Invitrogen Neon Transfection System (10 μL tip, 1400 V, 35 ms single pulse). Single cell–derived clonal lines with IKZF1 frameshift mutations in one or both alleles were identified by using a combined approach of bidirectional Sanger sequencing of an amplicon of ∼500 bp surrounding the Cas9 cleavage site and the TIDE (Tracking of Indels by Decomposition) algorithm prediction (https://tide.nki.nl).29 Clones with an insertion/deletion (indel) length of multiples of 3 were excluded as in-frame mutations. Clones with >2 or <2 unique, overlapping Sanger traces and/or abnormal TIDE predictions were rejected as containing possible copy number variations. Further confirmation of frameshift mutations was obtained by analysis of RNA sequencing (RNAseq) reads in the region surrounding the Cas9 cleavage site. In all cell lines, ablation of protein expression was confirmed by immunoblotting. Quantitative reverse transcription polymerase chain reaction (qRT-PCR) was also performed by using a probe set from downstream exons 7-8 to examine the effect of IKAROS protein ablation on messenger RNA (mRNA) levels.
Custom double-stranded DNA HDR templates were synthesized by Twist Biosciences (complete sequences are presented in the supplemental Methods). Clonal knock-in cell lines were generated by the same method, with the addition of 500-ng HDR template DNA to the sgRNA-Cas9 RNPs as previously described.30 For both the IK6–green fluorescent protein (GFP) fusion tag and the lentiviral GFP-transduced cell lines, GFP-positive cells were sorted on a BD FACS Aria III cytometer. Propidium iodide was used as dead cell exclusion stain. The plenti-CAG-IKZF1-V1-FLAG-IRES-GFP plasmid used in the rescue experiments was a gift from William Kaelin (Addgene plasmid # 107387).31 The pLenti-GIII-UbC-Puro SAMHD1 lentiviral vector was purchased from ABM (#LV295338, empty vector #LV589).
Statistical analysis
Statistical comparisons were evaluated by using the Student t test or one-way analysis of variance as indicated by using GraphPad Prism 7. All data are presented as mean ± standard error of the mean. For animal survival studies, Kaplan-Meier survival curves show time to morbidity and were analyzed by using the log-rank test. Where appropriate, exact P values are shown or indicated by a range of asterisks defined in the figure legends, and n indicates the number of biological replicates within each group.
Animal procedures were approved by the Institutional Animal Care and Use Committee of Baylor College of Medicine.
Additional information is provided in the supplemental Methods.
Results
Electroporation delivery of Cas9/sgRNA RNPs efficiently ablates IKAROS expression in human B-ALL cell lines
To identify critical transcriptional network alterations that could relate to changes in cell growth, signaling, and chemotherapy response, we engineered a set of clonal cell lines with accurately determined reduction or complete ablation of IKAROS protein expression. First, we electroporated the Nalm-6 B-ALL cell line (IKZF1 WT with DUX4 rearrangement and intragenic ERG deletion; IKZF1 mutations are observed in ∼20% of B-ALL patients with DUX4 rearrangements) with recombinant Cas9 protein precomplexed with sgRNAs (RNPs) targeting the first coding exons of IKZF1 (Figure 1A). To ensure that the electroporation conditions were optimized, as a control we used an sgRNA against CXCR4 that encodes a cell surface adhesion protein. After 72 hours, 59.2% of Nalm-6 cells electroporated with Cas9-CXCR4 sgRNA RNP exhibited ablation of cell surface expression of CXCR4, confirming efficient delivery of Cas9/sgRNA (Figure 1B). Sanger sequencing of DNA from the electroporated cell pools of PCR amplicons surrounding the IKZF1 target sites confirmed the high occurrence of indels, particularly for 3 of the 6 sgRNAs tested (sg55, sg57, and sg79), and all further experiments were performed with these 3 sgRNAs. After significant optimization, the Nalm-6 cell line proved amenable to manipulation using this electroporation system; it was therefore useful in many further experiments, in part due to its sensitivity to a variety of chemotherapeutic agents in culture and feasibility for in vivo transplantation.
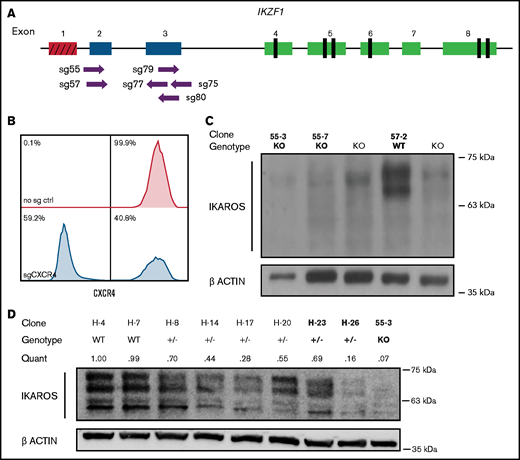
Cas9-sgRNA RNP targeting of IKZF1 efficiently eliminates protein expression in Nalm-6 cells. (A) sgRNAs targeting the early coding exons of IKZF1 (targeted exons 2 and 3, shown in blue; red indicates noncoding exon 1). Vertical black rectangles denote zinc fingers. (B) Flow cytometry histograms showing CXCR4 cell surface protein using sgRNA against CXCR4 as a control marker for CRISPR efficiency in Nalm-6. (C) IKAROS immunoblotting of single cell–derived Nalm-6 clones with Sanger sequencing–verified IKZF1 frameshift mutations. (D) Reduced-concentration RNP delivery yields a higher frequency of heterozygous clones with varying protein levels. Relative quantification was normalized to ACTIN. Bold font of clones on panels C and D indicates those used in further experiments.
Cas9-sgRNA RNP targeting of IKZF1 efficiently eliminates protein expression in Nalm-6 cells. (A) sgRNAs targeting the early coding exons of IKZF1 (targeted exons 2 and 3, shown in blue; red indicates noncoding exon 1). Vertical black rectangles denote zinc fingers. (B) Flow cytometry histograms showing CXCR4 cell surface protein using sgRNA against CXCR4 as a control marker for CRISPR efficiency in Nalm-6. (C) IKAROS immunoblotting of single cell–derived Nalm-6 clones with Sanger sequencing–verified IKZF1 frameshift mutations. (D) Reduced-concentration RNP delivery yields a higher frequency of heterozygous clones with varying protein levels. Relative quantification was normalized to ACTIN. Bold font of clones on panels C and D indicates those used in further experiments.
We derived and sequenced clonal cell lines and inferred their suspected indels from TIDE analysis. For subsequent experiments, clonal lines with complete ablation of IKAROS protein expression were selected (Figure 1C); these are referred to throughout as IKZF1 KO. Our initial approach generated no predicted heterozygous samples; 74% of edited clones sequenced had indels on both alleles (data not shown). To engineer clones with monoallelic IKZF1 lesions, we decreased the amount of Cas9 protein and sgRNA by 50% to 75% and successfully isolated clones with monoallelic, out-of-frame indels from these conditions, with reduced IKAROS protein expression ranging from 16% to 70% of WT expression levels (Figure 1D); these are hereafter referred to as IKZF1+/−. We selected as controls clonal lines with no evidence of IKZF1 editing by Sanger sequencing and expected levels of IKAROS protein expression, referred to throughout the text as IKZF1 WT. IKAROS protein is known to regulate its own expression level,32 and therefore, as expected, knockout clones showed a 3- to 4-fold upregulation of frameshift mutant IKZF1 mRNA by qRT-PCR (supplemental Figure 1), which served as a further surrogate confirmation of IKZF1 ablation.
To generate a corroborating set of orthogonal data, we also created a similar panel of heterozygous and knockout clones using the IKZF1 WT Tanoue B-ALL cell line [t(8;14) with IGH-MYC fusion] and confirmed loss of IKAROS protein (supplemental Figure 2A).
IKZF1 deletion results in a stem-like expression signature and upregulates targetable pathways
To identify the spectrum of gene expression changes in response to loss of IKAROS, we performed RNAseq on our clonal cell lines. Gene set enrichment analysis of the significantly upregulated or downregulated genes in KO clones showed a strong enrichment of a hematopoietic stem cell expression signature (consistent with previously published murine models)11 and an acute myeloid leukemia (AML) stem cell expression signature (Figure 2A). In addition, genes associated with the IL-7/JAK/STAT pathway were significantly altered between WT and KO cells, consistent with reported trends in expression profiling of ALL patient samples.12 The RNAseq reads spanning IKZF1 exon 2 were also used to confirm the CRISPR/Cas9–induced indels (supplemental Figure 3). We selected a subset of previously published IKAROS target genes33 for validation of the RNAseq results by qRT-PCR and found similar trends in gene expression changes using both methods (supplemental Figure 4).
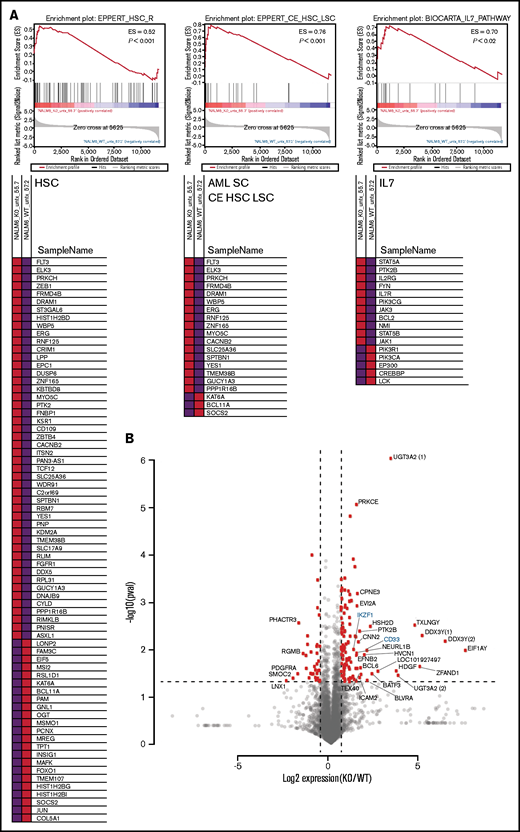
RNAseq analysis after IKAROS depletion reveals a stem cell–like gene expression pattern and upregulation of AML stem cell–associated genes. (A) Gene set enrichment analysis shows gene expression changes in signatures associated with hematopoietic stem cells (HSC), AML leukemia stem cells (AML SC CE HSC LSC), and the IL7/JAK/STAT pathways after IKZF1 knockout. Data shown are a representative example from a single pairwise gene set enrichment analysis of one WT and one IKZF1 KO experiment from 3 replicates. (B) Volcano plot showing the most statistically significantly upregulated and downregulated genes (IKZF1 KO vs WT). Parenthetical numerals next to gene identifiers indicate multiple isoforms of one gene. Genes highlighted in blue are included in confirmatory follow-up in other figures. ES, enrichment score.
RNAseq analysis after IKAROS depletion reveals a stem cell–like gene expression pattern and upregulation of AML stem cell–associated genes. (A) Gene set enrichment analysis shows gene expression changes in signatures associated with hematopoietic stem cells (HSC), AML leukemia stem cells (AML SC CE HSC LSC), and the IL7/JAK/STAT pathways after IKZF1 knockout. Data shown are a representative example from a single pairwise gene set enrichment analysis of one WT and one IKZF1 KO experiment from 3 replicates. (B) Volcano plot showing the most statistically significantly upregulated and downregulated genes (IKZF1 KO vs WT). Parenthetical numerals next to gene identifiers indicate multiple isoforms of one gene. Genes highlighted in blue are included in confirmatory follow-up in other figures. ES, enrichment score.
We noted that a number of the genes most downregulated after IKZF1 KO (eg, PDGFR) are involved in regulation of various cell cycle–dependent processes (Figure 2B); we thus performed cell cycle analysis in our edited Nalm-6 cells, finding a higher proportion of KO cells in the G1 phase in untreated samples (supplemental Figure 5A). These results suggested a relatively quiescent state, which aligns with the identified stem cell–like gene expression signature. WST-1 assays also revealed a substantially lower baseline metabolic rate in KO cells (supplemental Figure 5B). To track the relative growth rate of the B-ALL cells, we transduced our isogenic lines with a lentiviral GFP expression system and conducted an in vitro competition assay with WT + Lenti GFP vs KO, and the WT gradually outgrew the KO over 10 to 12 days (supplemental Figure 5C-D). These in vitro data suggest that IKZF1 deletion promotes not only a stem cell–like gene signature but also a stem cell–like phenotype.
IKZF1 deletion enhances bone marrow engraftment and shortens survival in mouse xenograft models
To determine the impact of IKAROS loss on bone marrow homing and engraftment of human B-ALL, we transplanted NSG mice intravenously with either the IKZF1 KO or WT Nalm-6 clones. Peripheral blood sampling on day 18, before noticeable signs of disease, showed a significantly higher engraftment in the animals transplanted with IKZF1 KO cells compared with WT (Figure 3A). Mice harboring IKZF1 KO B-ALL cells died of leukemia significantly earlier than the WT group (Figure 3B). Approximately 67% of mice transplanted with WT cells developed a mass at the retro-orbital injection site concurrent with development of systemic leukemia, but a similar phenomenon was only seen in 8% of KO-transplanted mice, suggesting significant adhesion-related changes that affected engraftment dynamics and tissue-specific tropism. These findings are consistent with the known increased expression of cell surface proteins responsible for adhesion in response to IKAROS disruption.18 At the time of euthanasia, IKZF1 KO xenografted animals displayed trends toward higher engraftment compared with WT in bone marrow and spleen, but a statistically significant difference was only observed in liver (Figure 3C). Similar patterns were seen in orthogonal experiments with IKZF1-deleted Tanoue B-ALL cell xenografts (supplemental Figure 2B), a highly aggressive in vivo model of B-ALL, which often results in moribund leukemic disease 1 to 2 weeks earlier than mice transplanted with Nalm-6 cells. Together, these results suggest that deletion of IKZF1 leads to more efficient motility and homing of B-ALL to the bone marrow and a more rapidly progressing, infiltrative disease.

IKZF1 deletion reduces survival in NSG mice and leads to profound dexamethasone resistance that is rescuable by IK1 overexpression. NSG mice xenografts transplanted with Nalm-6 clonal cell lines. (A) Flow cytometry for human HLA-ABC marker in peripheral blood samples taken on day 18. Horizontal bar shows mean, and error bars are standard error of the mean (SEM). Statistical significance was calculated by using the Student t test. (B) Kaplan-Meier survival curve; n = total number of mice in each group. Statistical significance was calculated by using the log-rank test. (C) Tissue engraftment of Nalm-6 cells in moribund mice at time of euthanasia. Horizontal bar shows mean, and error bars are SEM. Statistical significance was calculated by using the Student t test. (D, E, and G) Seventy-two-hour dexamethasone treatment of Nalm-6 clonal cell lines showing percent annexin V/7-AAD double-negative population as measured by flow cytometry. Samples were performed in triplicate; error bars show SEM. Asterisks denote statistical significance at the specified dose of KO compared with WT control (*P < .05; **P < .01; ***P < .001). (F) IKAROS immunoblotting of single cell–derived Nalm-6 clones and IK1 lentivirus overexpression (55-7 + IK1 rescue shown in purple). (G) Partial restoration of dexamethasone sensitivity in IKZF1 KO after IK1 rescue. (H) RNAseq gene expression levels plotted as fragments per kilobase of transcript per million mapped reads (FPKM) in untreated samples (NR3C1) or fold increase of FPKM after 48-hour, 50-nM dexamethasone treatment (FKBP5 and TSC22D3) (supplemental Figure 6C). BM, bone marrow; LIV, liver; ns, not significant; PB, peripheral blood; PBS, phosphate-buffered saline; SPL, spleen.
IKZF1 deletion reduces survival in NSG mice and leads to profound dexamethasone resistance that is rescuable by IK1 overexpression. NSG mice xenografts transplanted with Nalm-6 clonal cell lines. (A) Flow cytometry for human HLA-ABC marker in peripheral blood samples taken on day 18. Horizontal bar shows mean, and error bars are standard error of the mean (SEM). Statistical significance was calculated by using the Student t test. (B) Kaplan-Meier survival curve; n = total number of mice in each group. Statistical significance was calculated by using the log-rank test. (C) Tissue engraftment of Nalm-6 cells in moribund mice at time of euthanasia. Horizontal bar shows mean, and error bars are SEM. Statistical significance was calculated by using the Student t test. (D, E, and G) Seventy-two-hour dexamethasone treatment of Nalm-6 clonal cell lines showing percent annexin V/7-AAD double-negative population as measured by flow cytometry. Samples were performed in triplicate; error bars show SEM. Asterisks denote statistical significance at the specified dose of KO compared with WT control (*P < .05; **P < .01; ***P < .001). (F) IKAROS immunoblotting of single cell–derived Nalm-6 clones and IK1 lentivirus overexpression (55-7 + IK1 rescue shown in purple). (G) Partial restoration of dexamethasone sensitivity in IKZF1 KO after IK1 rescue. (H) RNAseq gene expression levels plotted as fragments per kilobase of transcript per million mapped reads (FPKM) in untreated samples (NR3C1) or fold increase of FPKM after 48-hour, 50-nM dexamethasone treatment (FKBP5 and TSC22D3) (supplemental Figure 6C). BM, bone marrow; LIV, liver; ns, not significant; PB, peripheral blood; PBS, phosphate-buffered saline; SPL, spleen.
IKZF1 deletion confers B-ALL with profound resistance to glucocorticoids by blunting response of glucocorticoid receptor target genes
We treated our IKZF1 KO and WT Nalm-6 clonal cell lines with increasing doses of the glucocorticoid (GC) dexamethasone. Although the IKZF1 WT cells underwent a dose- and time-dependent inhibition of proliferation and induction of apoptosis, the IKZF1 KO cells were highly resistant to these GC-induced effects (Figure 3D; supplemental Figure 6A). The heterozygous clone H-26 with ∼16% protein expression compared with WT (Figure 1D) displayed an intermediate resistance (Figure 3E), consistent with previously published findings using RNAi.33 Another heterozygous clone (H-23) with ∼69% of WT protein expression level showed no change in resistance. We observed similar results in orthogonal data generated with the clonal Tanoue cell lines (supplemental Figure 2C).
To further substantiate the claim that the observed drug resistance phenotypes are a direct result of IKZF1 depletion, the full-length protein isoform IK1 was introduced into the 55-7 Nalm-6 IKZF1 KO cell line using a bicistronic GFP lentiviral overexpression system. After sorting for GFP-positive cells, we confirmed IK1 overexpression (Figure 3F). When treated with dexamethasone, a partial rescue of GC sensitivity was observed in the IK1 overexpressing cells (Figure 3G), confirming IKAROS depletion as the driver of resistance.
RNAseq was also performed on Nalm-6 WT and IKZF1 KO clones after dexamethasone treatment. We selected the 50 nM dose at both the 24- and 48-hour time points because there was a small but detectable difference in apoptosis between WT and KO, while minimizing bias from inclusion of RNA from apoptotic cells (supplemental Figure 6B).34 In the KO cells, we observed lower baseline expression of NR3C1, the gene encoding the GC receptor, and a blunted response of several downstream GC receptor transcriptional target genes, including FKBP5 (a prolyl isomerase modulator of GC receptor sensitivity) and TSC22D3 (an anti-inflammatory, GC-induced leucine zipper); these results are consistent with previous findings (Figure 3H; supplemental Figure 6C).15,33 These data provide direct evidence for IKZF1 KO-driven GC resistance.
IKZF1 modulation alters sensitivity to chemotherapies in vitro
To test whether the observed effect was unique to GCs, we examined the impact of loss of IKAROS expression on sensitivity to a variety of chemotherapeutic agents used for the treatment of B-ALL. We found that full and partial ablation of IKAROS in Nalm-6 resulted in relative resistance to daunorubicin (Figure 4A; supplemental Table 1 [includes a summary of all 50% inhibitory concentration (IC50) values]) and asparaginase (Figure 4B). No statistically significant difference in sensitivity to methotrexate was observed (Figure 4C). The IKZF1 KO Tanoue cells displayed moderate resistance to vincristine and asparaginase in similar orthogonal experiments (supplemental Figure 2D-E).
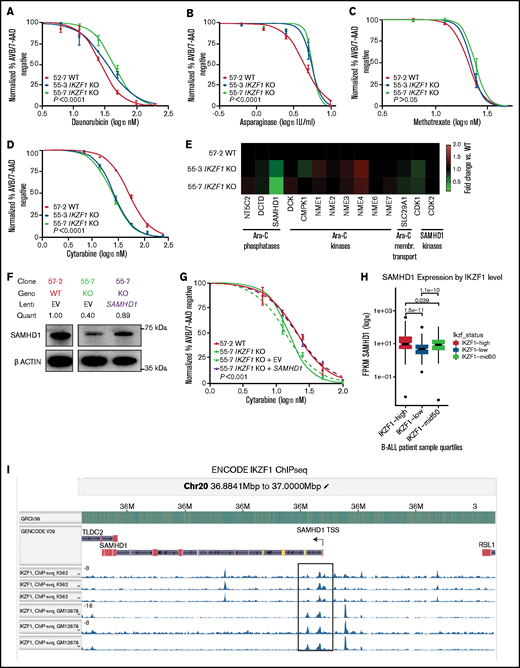
Deletion of IKZF1 leads to broadly increased chemoresistance. (A-D) and (G) Drug treatment of Nalm-6 clonal cell lines showing percent annexin V/7-AAD double-negative population as measured by flow cytometry. Samples were performed in triplicate; error bars show standard error of the mean, and statistical significance was calculated by using one-way analysis of variance. All assays were for 72 hours. Daunorubicin (A), asparaginase (B), methotrexate (no statistically significant difference) (C), and cytarabine (D). (E) Heatmap showing fold change of fragments per kilobase of transcript per million mapped reads (FPKM) of genes involved in cytarabine (Ara-C) enzymatic processing and membrane transport. Each data point shows the mean of 2 technical replicates. Fold changes are compared with WT sample mean. (F) western blot of SAMHD1 protein expression with lentivirally infected empty vector (EV) or ectopic SAMHD1 expressing constructs. Blot shown is a representative example from 3 experiments. (G) Cytarabine treatment with lentivirally transduced Nalm-6 cells. (H) SAMHD1 expression correlates with IKZF1 expression in B-ALL patient samples (PeCan database; n = 567 diagnostic RNAseq samples). (I) SAMHD1 locus from IKAROS chromatin immunoprecipitation sequencing (ChIPseq) from the ENCODE database, showing K562 and GM12878 (Epstein-Barr virus–transformed lymphoblastoid) cell lines.
Deletion of IKZF1 leads to broadly increased chemoresistance. (A-D) and (G) Drug treatment of Nalm-6 clonal cell lines showing percent annexin V/7-AAD double-negative population as measured by flow cytometry. Samples were performed in triplicate; error bars show standard error of the mean, and statistical significance was calculated by using one-way analysis of variance. All assays were for 72 hours. Daunorubicin (A), asparaginase (B), methotrexate (no statistically significant difference) (C), and cytarabine (D). (E) Heatmap showing fold change of fragments per kilobase of transcript per million mapped reads (FPKM) of genes involved in cytarabine (Ara-C) enzymatic processing and membrane transport. Each data point shows the mean of 2 technical replicates. Fold changes are compared with WT sample mean. (F) western blot of SAMHD1 protein expression with lentivirally infected empty vector (EV) or ectopic SAMHD1 expressing constructs. Blot shown is a representative example from 3 experiments. (G) Cytarabine treatment with lentivirally transduced Nalm-6 cells. (H) SAMHD1 expression correlates with IKZF1 expression in B-ALL patient samples (PeCan database; n = 567 diagnostic RNAseq samples). (I) SAMHD1 locus from IKAROS chromatin immunoprecipitation sequencing (ChIPseq) from the ENCODE database, showing K562 and GM12878 (Epstein-Barr virus–transformed lymphoblastoid) cell lines.
Interestingly, when treated with the nucleoside analogue cytarabine, the IKZF1 KO (IC50 11.62, 17.49 nM) and heterozygous cells were more sensitive than WT (23.07 nM) (Figure 4D; supplemental Table 1; supplemental Figure 7), suggesting that nucleoside analogues may be a particularly effective class of drugs for IKZF1-deleted B-ALL. Cytarabine is a cytosine analogue prodrug that must be sequentially triphosphorylated to its active form to be incorporated into DNA to produce a cytotoxic effect.35,36 Using our RNAseq results, we investigated the possibility of differential regulation of genes known to be involved in the processing of cytarabine and found only one potential candidate with significantly altered gene expression after IKZF1 KO, SAMHD1 (Figure 4E). SAMHD1 mRNA and protein expression were significantly lower in both IKZF1 KO clones relative to WT (supplemental Figure 8). A SAMHD1 lentiviral expression system with a relatively weak ubiquitin C promoter was used to restore SAMHD1 mRNA levels and SAMHD1 protein to near-WT levels (Figure 4F). We found that this ectopic SAMHD1 expression in IKZF1 KO shifted cytarabine sensitivity to that of the WT cells (IC50 [nM]: WT, 20.0; KO, 13.94; KO + empty vector, 15.31; KO + SAMHD1, 20.2) (Figure 4G).
We mined the publicly available St. Jude Cloud PeCan data portal (https://www.stjude.cloud)37 for SAMHD1 expression relative to IKZF1 expression in B-ALL samples. This data set includes 643 pediatric B-ALL samples with RNAseq data available. We excluded relapse cases, leaving 567 diagnostic B-ALL samples. A highly significant correlation was found, with samples in the lowest quartile of IKZF1 expression having the lowest SAMHD1 expression and samples with the highest quartile of IKZF1 expression having the highest SAMHD1 expression (Figure 4H). Examination of chromatin immunoprecipitation–sequencing data from the ENCODE database (https://www.encodeproject.org) revealed distinct IKAROS binding peaks near the promoter region of the SAMHD1 locus, indicating it may be a direct target of IKAROS regulation (Figure 4I). These data support that IKZF1 regulates expression of SAMHD1 in B-ALL.
GFP-tagged IK6 knock-in to the endogenous locus mimics the intragenic deletion found in patients
Although most clinical B-ALL trials group all IKFZ1 lesions together, some have found that different lesions have distinct effects on outcome. In particular, studies examining the prognostic impact of the IK6-generating intragenic deletion of exons 4-7 have been conflicting.19–21,38–42 Furthermore, in addition to inhibition of residual WT IKAROS, the IK6 isoform also heterodimerizes with other IKAROS family proteins such as HELIOS (encoded by IKZF2) and AIOLOS (encoded by IKZF3), inhibiting their function. Therefore, the transcriptomic and phenotypic impact of IK6 could differ from deletion of the entire IKZF1 gene. We thus expanded our panel of IKZF1 alterations with the creation of a novel IK6 dominant-negative isoform expressed under the control of the endogenous IKZF1 locus.
We used an HDR strategy in which exon 3 was directly fused to exon 8 on a 3.2 kb linear double-stranded DNA template, with an enhanced GFP fusion tag on the C terminus, serving the dual purpose of selection by cell sorting and tracking of intracellular localization (Figure 5A). This strategy successfully generated GFP-expressing cells (efficiencies of 0.1%-0.4% of live cells) (Figure 5B). GFP-positive cells were sorted, one cell per well, into 96-well plates. After expansion, clones were selected based on PCR and Sanger sequencing that had one WT unedited allele, with the other allele showing perfect homologous recombination matching the template sequence.

Creation and characterization of IK6-expressing B-ALL via knock-in of IK6 into the endogenous IKZF1 locus. (A) HDR strategy for GFP-fused IK6 knock-in using 3.2 kb double-stranded DNA HDR template. Targeted exon 3 shown in purple; red indicates noncoding exon 1. *Mutated protospacer-adjacent motif (PAM) to prevent cleavage of template; vertical black rectangles denote zinc fingers. (B) Flow cytometry plots showing GFP+ sort gate used to purify cells that had IK6-GFP knock-in. (C) IK6-GFP fusion protein in nucleus and mislocalization to cytoplasm as shown by live cell confocal microscopy. Upper row, Nalm-6 GFP-positive pool showing heterogeneous expression 1 month after sorting; lower row, Reh single cell–derived clone. All images were taken on the GE Healthcare DeltaVision LIVE High Resolution Deconvolution Microscope using an Olympus 40X U Apo/1.35 NA oil objective with iris and analyzed with SoftWoRx. Scale bars are 20 µm. (D) Immunoblotting for IKAROS and GFP-IK6 fusion protein in Nalm-6 and Reh single cell–derived clones. (E) In vitro competition of Nalm-6 (upper graph) and Reh (lower graph). GFP was measured by flow cytometry at indicated time points. All samples were performed in triplicate, and error bars show standard error of the mean. Additional details are provided in supplemental Figure 5C-D.
Creation and characterization of IK6-expressing B-ALL via knock-in of IK6 into the endogenous IKZF1 locus. (A) HDR strategy for GFP-fused IK6 knock-in using 3.2 kb double-stranded DNA HDR template. Targeted exon 3 shown in purple; red indicates noncoding exon 1. *Mutated protospacer-adjacent motif (PAM) to prevent cleavage of template; vertical black rectangles denote zinc fingers. (B) Flow cytometry plots showing GFP+ sort gate used to purify cells that had IK6-GFP knock-in. (C) IK6-GFP fusion protein in nucleus and mislocalization to cytoplasm as shown by live cell confocal microscopy. Upper row, Nalm-6 GFP-positive pool showing heterogeneous expression 1 month after sorting; lower row, Reh single cell–derived clone. All images were taken on the GE Healthcare DeltaVision LIVE High Resolution Deconvolution Microscope using an Olympus 40X U Apo/1.35 NA oil objective with iris and analyzed with SoftWoRx. Scale bars are 20 µm. (D) Immunoblotting for IKAROS and GFP-IK6 fusion protein in Nalm-6 and Reh single cell–derived clones. (E) In vitro competition of Nalm-6 (upper graph) and Reh (lower graph). GFP was measured by flow cytometry at indicated time points. All samples were performed in triplicate, and error bars show standard error of the mean. Additional details are provided in supplemental Figure 5C-D.
We observed the IK6-GFP fusion protein, as expected,43 partially aberrantly localized to the cytoplasm in Nalm-6 cells, and similar orthogonal data were also generated by using the Reh cell line [an IKZF1 WT B-ALL with t(12;21) ETV6-RUNX1 fusion] (Figure 5C). Immunoblot of whole cell extracts showed that IK6 was more highly expressed than the normal IK1/IK2 isoforms found in the WT clone (Figure 5D). This phenomenon is commonly seen in cell lines and patient samples expressing IK6, presumably owing in part to reduced degradation secondary to lack of a ubiquitin ligase-interacting domain.44 Both the Nalm-6 and Reh WT/IK6 cells proliferated less than WT over several weeks of coculture, consistent with the IKZF1 KO cell lines and suggestive of a relatively quiescent, stem cell–like phenotype (Figure 5E). Thus, with our CRISPR/Cas9 HDR strategy, we successfully generated B-ALL cells with expected IK6 expression and cellular localization, ideal for direct comparison with IKZF1 WT, KO, and +/− to precisely delineate the impact of these various IKZF1 genotypes commonly encountered in human B-ALL.
Because the GFP tag in our engineered IK6-expressing cells is near the protein-binding zinc finger domain, it is possible that the GFP tag could interfere with protein function. To explore this possibility, we generated isogenic Nalm-6 cell lines with a GFP tag on the C terminus of one allele of the WT IKZF1 using a CRISPR/Cas9 HDR strategy similar to that used to generate the IK6-GFP cell lines (supplemental Figure 9A). We found that the WT IKAROS-GFP fusion protein predominantly localized to the nucleus and with lower expression than the IK6-GFP, as expected (supplemental Figure 9B-C). These cells were also nearly identical to IKZF1 WT cells without a GFP tag in analyses of RNA and protein expression and drug sensitivity (supplemental Figure 9D-F). These investigations suggest that the GFP fused to IKAROS proteins minimally affects cellular phenotype and function.
Impact of IKZF1 genotype on gene expression
To assess gene expression changes induced by the different IKZF1 genotypes, we performed RNAseq on our engineered Nalm-6 cell lines, comparing IKZF1 WT, KO, +/−, and IK6-GFP/WT cells. Isogenic lines of the same IKZF1 genotype had similar changes in gene expression (Figure 6A). In general, the differentially expressed genes were similarly expressed across all the IKZF1 mutated lines, with the most pronounced effects in most genes noted in the IKZF1 KO (Figure 6A-B). The findings were validated at the protein level by flow cytometry of several clinically relevant, cell surface–expressed phenotyping markers, including CD19, CD22, CD33, and FLT3 (Figure 6C). CD33 was highly upregulated in KO cells, with a more modest increase in both +/− and IK6-GFP/WT lines. CD33 expression was similar in WT and WT IKZF1-GFP cells (supplemental Figure 9E). CD22 and CD19 also increased with all IKZF1 lesions, although the effect was less pronounced in the KOs than in IK6-GFP/WT. FLT3 was also modestly upregulated in the KO cells; however, it was not expressed on the cell surface in the parental cell line. Overall, B-ALL IKZF1 lesions result in a similar gene expression pattern, with the magnitude of gene expression changes influenced largely by the amount of residual IKAROS function.
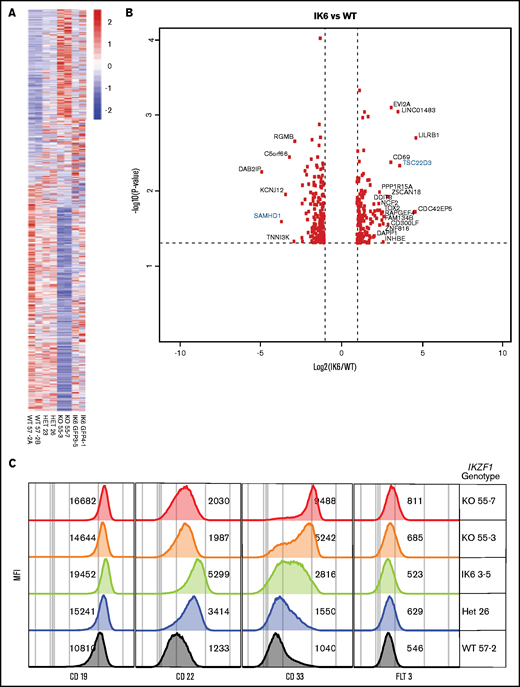
IK6 knock-in shows changes in gene expression patterns similar to heterozygous knockout. All samples are from Nalm-6 clonal cell lines. (A) Heatmap showing differential gene expression analysis from RNAseq from WT, heterozygous (HET), KO, and IK6 knock-in. (B) Volcano plot highlighting the most significantly upregulated or downregulated genes (IK6 vs WT). Genes highlighted in blue are included in confirmatory follow-up. (C) Flow cytometry histograms showing confirmation of protein expression changes of a panel of cell surface markers commonly used in leukemia phenotyping. MFI, mean fluorescence intensity.
IK6 knock-in shows changes in gene expression patterns similar to heterozygous knockout. All samples are from Nalm-6 clonal cell lines. (A) Heatmap showing differential gene expression analysis from RNAseq from WT, heterozygous (HET), KO, and IK6 knock-in. (B) Volcano plot highlighting the most significantly upregulated or downregulated genes (IK6 vs WT). Genes highlighted in blue are included in confirmatory follow-up. (C) Flow cytometry histograms showing confirmation of protein expression changes of a panel of cell surface markers commonly used in leukemia phenotyping. MFI, mean fluorescence intensity.
IK6 knock-in transplanted NSG mice have accelerated disease progression compared with WT
To establish the impact of IK6 expression on bone marrow homing and engraftment, we repeated the NSG mouse xenograft experiment with the Nalm-6 WT/IK6 cells compared with WT and KO counterparts. Peripheral blood engraftment of WT/IK6 Nalm-6 cells 2 weeks posttransplant was similar to WT, whereas KO engraftment was significantly higher, as in our previous experiments (Figure 7A). Despite this lower early peripheral blood engraftment, mice transplanted with IK6 knock-in cells died of the disease at approximately the same time as the KO transplanted animals (Figure 7B). Liver weight was significantly increased in the IK6 transplanted mice (Figure 7C), but spleen weight and bone marrow engraftment were not significantly different (supplemental Figure 10). These results suggest that, although IK6 was associated with increased liver infiltration with less early peripheral blood engraftment, IK6 and KO contribute similarly to overall B-ALL disease progression.
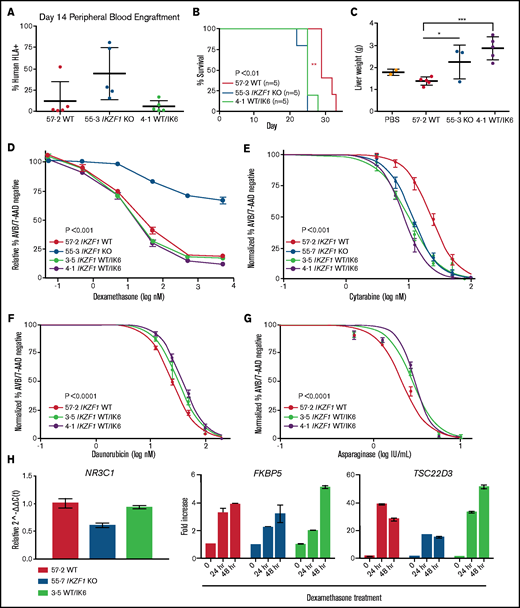
IK6 knock-in leads to shortened survival time in NSG mice with increased extramedullary organ engraftment. NSG mice xenografts transplanted with Nalm-6 clonal cell lines. (A) Flow cytometry for human HLA-ABC marker in peripheral blood samples taken on day 14. (B) Kaplan-Meier survival curve; n = total number of mice in each group. Statistical significance was calculated by using one-way analysis of variance. (C) Mouse liver weights at time of euthanasia. Statistical significance was calculated by using the Student t test. Drug treatment of Nalm-6 clonal cell lines showing percent annexin V/7-AAD double-negative population as measured by flow cytometry after 72 hours of treatment with dexamethasone (D), cytarabine (E), daunorubicin (F), and asparaginase (G). (H) TaqMan qRT-PCR analysis of relative expression of NR3C1 at baseline and fold change in expression of FKBP5 and TSC22D3 after dexamethasone treatment in Nalm-6 clonal cell lines of indicated genotype. For all experiments, samples were performed in technical triplicate. Error bars show standard error of the mean. *P < .05; **P < .01; ***P < .001. PBS, phosphate-buffered saline.
IK6 knock-in leads to shortened survival time in NSG mice with increased extramedullary organ engraftment. NSG mice xenografts transplanted with Nalm-6 clonal cell lines. (A) Flow cytometry for human HLA-ABC marker in peripheral blood samples taken on day 14. (B) Kaplan-Meier survival curve; n = total number of mice in each group. Statistical significance was calculated by using one-way analysis of variance. (C) Mouse liver weights at time of euthanasia. Statistical significance was calculated by using the Student t test. Drug treatment of Nalm-6 clonal cell lines showing percent annexin V/7-AAD double-negative population as measured by flow cytometry after 72 hours of treatment with dexamethasone (D), cytarabine (E), daunorubicin (F), and asparaginase (G). (H) TaqMan qRT-PCR analysis of relative expression of NR3C1 at baseline and fold change in expression of FKBP5 and TSC22D3 after dexamethasone treatment in Nalm-6 clonal cell lines of indicated genotype. For all experiments, samples were performed in technical triplicate. Error bars show standard error of the mean. *P < .05; **P < .01; ***P < .001. PBS, phosphate-buffered saline.
IK6 knock-in creates a unique drug resistance profile
To determine if B-ALL with IK6 expression had a different chemosensitivity profile compared with B-ALL with deletion of IKZF1, WT and IK6 cell lines were treated with chemotherapy. We observed no change in sensitivity to dexamethasone in IK6 cells compared with WT (Figure 7D). Also, as with the IKZF1 KO and IKZF1+/−, the WT/IK6 cells exhibited increased sensitivity to cytarabine (IC50 IK6, 8.65, 11.99; WT, 23.07 nM) (Figure 7E). Conversely, the WT/IK6 cells displayed moderate resistance to daunorubicin and asparaginase, similar to the IKZF1 KO and IKZF1+/− clones (Figure 7F-G), and the WT 57-2 clone was similar to the Nalm-6 parental cell line with all of these agents (supplemental Figure 11; supplemental Table 1).
To explore the mechanisms of retained GC sensitivity, we found that unlike the IKZF1 KO cells, IK6 cells had levels of NR3C1 expression similar to that of WT cells (Figure 7H). In addition, whereas IKZF1 KO cells have a blunted response of GC response genes after dexamethasone treatment, the expression of these response genes after dexamethasone treatment of IK6 cells was similar to that of WT cells. These data, combined with data from our Nalm-6 IKZF1+/− clones with varying levels of residual IKAROS protein, suggest that WT IKAROS protein levels must be severely reduced (to <20% of normal levels) in Nalm-6 cells to cause GC resistance.
Overall, these studies suggest that considering the specific type of IKZF1 lesions may inform treatment decisions.
Discussion
To detect small changes in chemoresistance that would have been previously undetectable using knockdown approaches, we generated complete IKAROS protein knockout in human cells. Using these engineered cell lines, we could confirm previous findings from various IKZF1 model systems and primary patient samples, such as stem cell–like gene expression pattern and increased JAK/STAT signaling, providing evidence that these novel model systems are valid tools with which to study the biology of IKZF1 in human B-ALL. Further work using these engineered cell lines showed that IKZF1 loss leads to relative cell quiescence, enhanced bone marrow engraftment, and resistance to multiple chemotherapeutic agents. These results are consistent with the phenotype observed in patients with IKZF1 deletions, which are associated with increased risk of relapse in clinical studies. To date, some cancer consortia have incorporated IKZF1 status along with bone marrow minimal residual disease into their risk stratification schema, defining patients with IKZF1 mutations/deletions and minimal residual disease–positive disease as high risk and assigning them to intensified postinduction therapy.45 Other published studies using varying methods of measurement and statistical analyses have shown, however, little benefit of IKZF1 status as a classifier in risk stratification,40 although these data are from a relatively small cohort (reviewed in Olsson and Johansson46 ). Overall, our studies show that lesions of IKZF1 result in cell-intrinsic resistance to most ALL-directed chemotherapy.
We identified patterns of chemotherapy sensitivity in IKZF1-mutant B-ALL that may have important clinical implications. Complete ablation of IKAROS protein expression in B-ALL cells resulted in profound dexamethasone resistance, consistent with prior work in other model systems. However, a different pattern was observed with other genotypes, with potential biologic implications. In the sequencing-confirmed IKZF1+/− isogeneic clones, we found that IKAROS protein expression varied, which allowed us to determine relative corticosteroid resistance on the basis of protein level. Interestingly, we only observed a measurable effect on dexamethasone sensitivity, mediated by decreased NR3C1 expression and the blunted expression of GC response genes after dexamethasone treatment, in clones with very low residual IKAROS levels (<20%). In addition, although the chemosensitivity profile of the WT/IK6 clones is similar to that of the IKZF1 KO clones, the WT/IK6 cells maintained their NR3C1 expression and sensitivity to dexamethasone. These findings suggest IKAROS is necessary for dexamethasone sensitivity with a threshold level of required protein expression, below which an IKAROS dose-dependent effect occurs. Of note, it is possible that the GFP tag on the IK6 isoform could affect protein–protein interactions, thus partially interfering with its dominant negative function. Although we believe this is unlikely to be the full explanation for the difference in GC sensitivity based on other functional studies performed on these cells, future work will include generation of an untagged IK6 isoform to confirm these findings.
Although all IKZF1 engineered lesions result in relative resistance to most standard ALL-directed agents, we found a uniform increased sensitivity to the nucleoside analogue cytarabine that seems to be a result of the reduced expression of SAMHD1, a dNTP/cytarabine-TP triphosphohydrolase,47 which could be fully rescued with restoration of WT SAMHD1 protein levels. Cell line and primary patient data further corroborate that IKAROS regulates SAMHD1 expression. Confirming its role in nucleoside analogue sensitivity, high SAMHD1 expression has been identified as an important potential biomarker in AML for cytarabine resistance.48 It is also implicated in hypomethylating agent resistance in AML,49 and pharmacologic inhibition of SAMHD1 with ribonucleotide reductase inhibitors such as hydroxyurea or gemcitabine can synergistically increase the efficacy of cytarabine in AML preclinical models.36 Although low-dose cytarabine is included in a subset of B-ALL postinduction therapy cassettes, our findings suggest that regimens with intensification of cytarabine may preferentially benefit patients with IKZF1-deleted B-ALL.
Comparing the gene expression profile and cell surface protein expression of our engineered B-ALL cell lines with and without IKZF1 deletion, we observed upregulation of the myeloid-associated cell surface phenotypic markers CD33 and FLT3 with IKAROS ablation, which may provide additional targeted therapeutic vulnerabilities. The CD33-targeted antibody-drug conjugate gemtuzumab ozogamicin has activity against CD33+B-ALL in vitro, in patient-derived xenografts,50 and has successfully induced complete remission in patients.51–53 We also observed increased cell surface expression of CD19 and CD22, indicating that deletions/mutations of IKZF1 should not detrimentally affect sensitivity to CD19- or CD22-targeted therapies such as blinatumomab and inotuzumab ozogamicin, or CD19 and CD22 targeting chimeric antigen receptor T cells. We also found upregulation of the potentially targetable JAK/STAT pathway with IKZF1 loss. Importantly, IKZF1 aberrations are highly prevalent in Philadelphia chromosome (Ph)-positive and Ph-like B-ALL, which are characterized by activation of signaling pathways due to gene fusions and/or mutations, including subsets with JAK/STAT-activating lesions. Notably, all our investigations were conducted in Ph-negative, non–Ph-like B-ALL cells. Despite the heterogeneity of different driver fusion translocations found in the various cell lines used, similar effects of IKZF1 manipulation were observed in vitro and in vivo. How IKZF1 loss affects signaling pathways in Ph-positive and Ph-like leukemia will be important future questions.
Overall, our data support clinical and laboratory studies reporting that IKZF1-mutated B-ALL is an aggressive, infiltrative, and treatment-resistant disease, indicating we can generate reliable model systems via CRISPR engineering of human cell lines. Investigations with these engineered cell lines indicate that detailed delineation of the exact IKZF1 status in patients with ALL at diagnosis may be informative in more accurately determining risk stratification and the most effective therapeutic regimen.
Acknowledgments
The authors thank the members of the Goodell Laboratory for helpful discussions. The authors also gratefully acknowledge editorial assistance with the manuscript from Catherine Gillespie, employed by the Department of Cell and Gene Therapy, Baylor College of Medicine.
The work was supported by the National Institutes of Health (NIH), National Cancer Institute (K08 CA 201611 and K12 CA090433-11), Be The Light Charitable Foundation, Golfers Against Cancer, The Gillson-Longenbaugh and Anderson Foundation, Hyundai Hope on Wheels, The Ladies Leukemia League, Rally Foundation for Childhood Cancer Research, Hope Versus Cancer Foundation (R.E.R.), American Society of Hematology HONORS Award (R.G.), and Alex's Lemonade Stand POST award (C.J.). All flow cytometry experiments were performed at the Baylor College of Medicine Cytometry and Cell Sorting Core, which is supported by the NIH, National Cancer Institute P30 Cancer Center Support (CA125123) the NIH, National Center for Research Resources (RR024574) and the NIH, Office of the Director (S10 OD025251). Microscopy experiments were performed at the Baylor College of Medicine Integrated Microscopy Core, which is supported by the NIH, National Cancer Institute P30 Cancer Center Support (CA125123), the NIH, National Institute of Diabetes and Digestive and Kidney Diseases (56338-13/15), the Cancer Prevention and Research Institute of Texas (Cancer Prevention and Research Institute of Texas (RP150578), and the John S. Dunn Gulf Coast Consortium for Chemical Genomics. STR DNA fingerprinting was done at The University of Texas MD Anderson Cancer Center CCSG-funded Characterized Cell Line Core, with support from the NIH, National Cancer Institute (CA016672).
Authorship
Contribution: J.H.R., R.G., A.G., M.C.G., G.M., R.A., T.S., C.J., S.B., C.D.D.C., S.E.C., K.K., L.B., and R.E.R. performed experiments; J.H.R., R.G., M.C.G., L.B., M.A.G., and R.E.R. designed experiments and analyzed experimental results; J.M.R. and M.C.G. performed bioinformatics analysis; and J.H.R. and R.E.R. wrote the manuscript; all authors reviewed and commented on the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Rachel E. Rau, Baylor College of Medicine, 1102 Bates Ave, Suite 1025, Houston, TX 77030; e-mail: rerau@bcm.edu.

