

Key Point
DKK1 binds CKAP4 receptor to activate NF-κB signaling rendering resistance to proteasome inhibitor treatment in MM cells.

Abstract
Proteasome inhibitors, such as bortezomib (BTZ), represent the key elements in chemotherapy regimens for multiple myeloma (MM), whereas acquired chemoresistance and ultimately relapse remain a major obstacle. In the current study, we screened differently expressed cytokines in bortezomib-resistant MM cells and found that Dickkopf-1 (DKK1) level was remarkably augmented, whereas CD138 level was significantly suppressed. DKK1 in vitro specifically enhanced the resistance of myeloma cells to bortezomib treatment, and excessive DKK1 drove CD138 downregulation via inhibition of canonical Wnt signaling. Notably, DKK1 mainly induced drug resistance in MM cells via the receptor of CKAP4. Mechanistically, CKAP4 transduced DKK1 signal and evoked NF-κB pathway through recruiting and preventing cullin associated and neddylation dissociated 1 from hampering the assembly of E3 ligase-mediated ubiquitination of IκBα. In addition, we found that interleukin-6 (IL-6) stimulated CKAP4 expression to generate drug resistance, and disturbance of DKK1-CKAP4 axis improved sensitivity to BTZ treatment of MM and attenuated bone destruction in a mouse model. Collectively, our study revealed the previously unidentified role of DKK1 in myeloma drug resistance via Wnt signaling dependent and independent manners, and clarified the importance of antagonism of DKK1-IL-6 loop in bone marrow microenvironment.
Introduction
Multiple myeloma (MM) is the second most prevalent hematologic neoplasm characterized primarily by monoclonal growth of terminally differentiated plasma cells in the bone marrow. The infiltration of MM cells and accumulation of monoclonal immunoglobulin protein give rise to the generation of destructive bone disease. As a consequence, patients with MM frequently suffer from severe complications, including anemia, renal failure, and osteolytic bone lesion.1,2 With the application of proteasome inhibitors (PIs), such as bortezomib (BTZ), the overall response rates, progression-free survival, and overall survival of myeloma patients have been all effectively improved over the past 15 years. Nonetheless, in parallel with the flourishing development of novel therapy regimens is the occurrence of acquired drug resistance, and inevitably the outcome of resistance to chemotherapy, rendering great challenges to the remedy of relapsed and refractory myeloma.3,4
This highlights the utmost importance to define the complexity of acquired drug resistance in MM. Several mechanisms contribute to the generation of PI resistance, including accelerated drug expulsion, deranged signaling pathways, genetic abnormalities and epigenetic aberrations, activation of endoplasmic reticulum stress, and microenvironment.5,6 Besides, bone marrow creates a permissive microenvironment to interact with MM plasma cells and influences disease development and drug resistance.7 Bone marrow stromal cells (BMSCs) offer outgrowth and survival signals through adhesion molecules and secretion of cytokines such as interleukin-6 (IL-6),8 which could augment the transcription of antiapoptotic MCL-1 in a STAT3-dependent manner.9 It is well known that MM cells secrete high levels of the Wnt inhibitor, Dickkopf-1 (DKK1), which mainly prevents BMSCs from differentiating into osteoblasts.10 The Munshi group evaluated the effect of targeting DKK1 using a neutralizing antibody, BHQ880, and found that anti-DKK1 significantly inhibited growth of MM cells in the presence of BMSCs in vitro, and this benefit was associated with inhibition of production of IL-6.11 Gunn et al reported that marrow stromal cell–conditioned media promoted the proliferation of DKK1-secreting MM cells and an IL-6–neutralizing antibody largely preluded this effect; therefore, interruption of targeting DKK1/IL-6 loop may suppress proliferation of MM cells and alleviate osteolytic lesions.12 These studies argue DKK1 as an important therapeutic target in MM and suggest a potential close relationship between DKK1 and IL-6. However, the underlying regulating machinery of DKK1 and IL-6 in MM cells, especially in modulating drug resistance, has not been investigated.
CKAP4, a type II transmembrane protein, was originally found as an endoplasmic reticulum membrane protein and anchors the endoplasmic reticulum to microtubules.13 Recently, CKAP4 was discovered to localize to the plasma membrane of some cells and identified as a novel DKK1 receptor.14,15 Indeed, DKK1 and CKAP4 expression was frequent in tumor lesions of human pancreatic, esophageal, and lung cancers, and simultaneous expression of both proteins in patient tumors was negatively correlated with prognosis and relapse-free survival.16 A neutralizing antibody against CKAP4 demonstrated the repressive activities for the binding of DKK1 and CKAP4, also blocked xenograft tumor formation in immunodeficient mice and extended the survival of mice receiving intraperitoneal or orthotopic injection of tumor cells.17,18 However, whether CKAP4 plays any role in MM malignancy and how CKAP4 ties DKK1 and IL-6 regulating loop has been totally unknown. In this study, we aim to evaluate the role of DKK1 in modulating sensitivity of MM cells to PI treatment, and illustrate the regulating mechanism of a new IL-6-CKAP4-DKK1 loop in MM cells.
Materials and Methods
Cell lines, cell proliferation, and flow cytometry assays
MM cell lines used in our laboratory and the induction of BTZ-resistant cells have been described in a previous publication.19 Cells were Short Tandem Repeat authenticated and substantiated as mycoplasma-free. Microscale thermophoresis assays to detect cell proliferation and flow cytometry to detect the apoptosis were performed as previously described.19,20
Immunoassays
Antibody-based immunoblotting has been described in our previous study.19 All antibodies, vendors, and dilutions are provided in the supplemental Methods. The representative western blot images for at least 3 independent experiments shown in the figures have been cropped and auto-contrasted. Quantification of western blot images and immunohistochemistry staining intensities were analyzed using the Image J 1.46r software for all samples in each group. All gray density values of western blot bands, either for a single band or the cleaved PARP, were corrected by β-actin or GAPDH loading control. Statistical analysis of all western blot images is shown in the supplemental Methods. Detailed protocols can be found in the supplemental Methods.
NOD/SCID xenograft and bone lesion mice models
Animal studies were approved by the Committee on Animal Research and Ethics of Tianjin Medical University, and all protocols conformed to the Guidelines for Ethical Conduct in the Care and Use of Nonhuman Animals in Research. Four- to 6-week-old female NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ (NSG) mice were used to establish the xenograft (n = 12) and intra-bone injection models (n = 8) as previously reported.20,21
Statistical analysis
Data were shown as mean ± standard deviation for at least 3 independent experiments. Differences between groups were determined using Student t test or 1-way analysis of variance. Pearson correlation test was used to determine the correlations between gene expressions, and survival analysis and a log-rank test was done by GraphPad Prism 8.0. A P value < .05 was considered statistically significant. *P ≤ .05; **P ≤ .01, ***P ≤ .01, compared with the controls, respectively. Detailed protocols can be found in the supplemental Methods.
Results
DKK1 enhances drug resistance to proteasome inhibitors of MM cells
To investigate the features and mechanisms of PI-induced drug resistance in MM cells, we established BTZ-resistant (BR) MM cell lines.19 Subsequently, we analyzed cytokine profiles in BR or counterpart (wild-type [WT]) MM.1S cells, and found BTZ resistance yielded 64 differently expressed cytokines using chips containing 440 cytokines (supplemental Table 1), in which 14 cytokines were obviously decreased and 50 remarkably increased (cutoff = 2, fold change) (Figure 1a). The Gene Ontology and Kyoto Encyclopedia of Genes and Genomes analysis of these results found enhanced cytokine-cytokine receptor interaction and activated NF-κB pathway in BR MM.1S cells (Figure 1B; supplemental Figure 1A). Interestingly, DKK1 was screened as one of the top increased cytokines, but CD138 was significantly suppressed in the BR cells, and western blot or enzyme-linked immunosorbent assay (ELISA) also confirmed DKK1 upregulation in BR cells (Figure 1C-D).
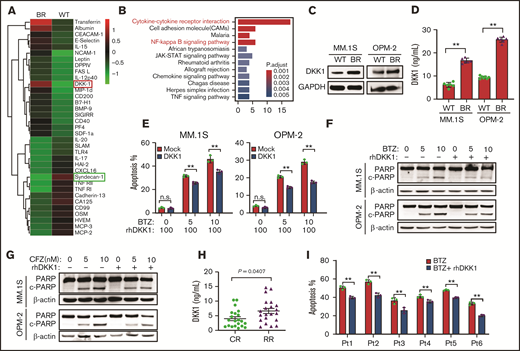
DKK1 promotes MM cell malignancy. (A) Heat map shows selected up- and downregulated cytokines of the WT and BR MM.1S cells. (B) Kyoto Encyclopedia of Genes and Genomes analysis highlights the upregulated or downregulated signaling pathways in the BR MM.1S cells. (C) Representative protein levels of DKK1 in WT and BR MM cells. (D) DKK1 levels in WT and BR MM.1S and OPM-2 cells measured by ELISA (n = 12). (E) Quantification of apoptotic MM cells treated with increasing dosage of BTZ (0-10 nM) with or without 100 ng/mL recombinant human DKK1 for 48 hours (n = 3). (F) Cleavage of PARP as the apoptotic marker in MM cells treated with or without rhDKK1 (100 ng/mL), combined with increasing dosage of BTZ and CFZ (G) for 24 hours (n = 3). (H) DKK1 levels in bone marrow plasma cell (BMPC) from patients with CR (n = 12) and RR patients RR (n = 12) measured by ELISA. (I) Quantification of apoptosis in CD138+ cells from 6 MM patients treated with BTZ (5 nM) with or without 100 ng/mL rhDKK1 for 12 hours.
DKK1 promotes MM cell malignancy. (A) Heat map shows selected up- and downregulated cytokines of the WT and BR MM.1S cells. (B) Kyoto Encyclopedia of Genes and Genomes analysis highlights the upregulated or downregulated signaling pathways in the BR MM.1S cells. (C) Representative protein levels of DKK1 in WT and BR MM cells. (D) DKK1 levels in WT and BR MM.1S and OPM-2 cells measured by ELISA (n = 12). (E) Quantification of apoptotic MM cells treated with increasing dosage of BTZ (0-10 nM) with or without 100 ng/mL recombinant human DKK1 for 48 hours (n = 3). (F) Cleavage of PARP as the apoptotic marker in MM cells treated with or without rhDKK1 (100 ng/mL), combined with increasing dosage of BTZ and CFZ (G) for 24 hours (n = 3). (H) DKK1 levels in bone marrow plasma cell (BMPC) from patients with CR (n = 12) and RR patients RR (n = 12) measured by ELISA. (I) Quantification of apoptosis in CD138+ cells from 6 MM patients treated with BTZ (5 nM) with or without 100 ng/mL rhDKK1 for 12 hours.
To further ascertain the effects of DKK1 on the malignant phenotypes of MM cells, we treated MM cells with exogenous recombinant human DKK-1 (rhDKK1) in vitro. Cultured MM.1S and OPM-2 cells in presence of rhDKK1 exhibited similar proliferation ability compared with these in normal medium (supplemental Figure 1B), and similar finding was observed in cell cycle (supplemental Figure 1C-D). Intriguingly, when DKK1 was present in MM cells treated with BTZ or carfilzomib (CFZ), significant lower apoptosis rates were elicited compared with the control (supplemental Figure 2A; Figure 1E). The potentiated capability in cell survival offered by DKK1 was also confirmed by detecting the cleavage of PARP-1 by western blotting (Figure 1F-G). By comparison, DKK1 did not alter the drug sensitivity of MM cells to melphalan or dexamethasone (supplemental Figure 2B-C). Collectively, these results argue that DKK1 specifically enhances the resistance of myeloma cells to PIs.
Clinically, we examined DKK1 expression via ELISA in complete response (CR) and refractory/relapsed (RR) MM patients, and found DKK1 level was significantly elevated in RR MM patients rather than in CR patients (Figure 1H). Importantly, when DKK1 was in presence of BTZ treatment in 6 cases of CD138+ plasma cells from MM patients, significant lower apoptosis rates could be elicited compared with the controls (Figure 1I). These data indicated that high level of DKK1was associated with PI resistance in MM patients.
DKK1 suppresses CD138 expression via canonical Wnt/β-catenin signaling pathway
CD138 is a heparin sulfate proteoglycan responsible for myeloma malignancy, and myeloma cells expressing low levels of CD138 are believed to be an immature phenotype and resistant to lenalidomide.22 Because our cytokine array data indicated a negative correlation between DKK1 and CD138 in BR MM cells, we further investigated the effects of DKK1 on CD138 expression. After treated for 24 hours, DKK1 decreased the messenger RNA (mRNA) level of CD138 in a dose-dependent manner (Figure 2A), as well as downregulated CD138 protein level (Figure 2B). Notably, when CD138+ plasma cells from 6 cases of MM patients were treated with rhDKK1 for 12 hours, CD138 expression were all obviously suppressed (Figure 2C).
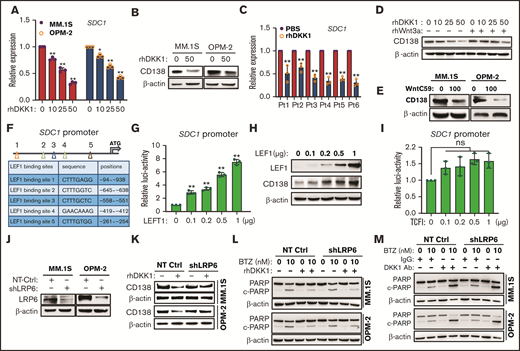
DKK1 maintains MM cell immatureness by downregulating CD138. (A) Quantitative PCR (qPCR) shows expressions of SDC1 (CD138) of MM.1S and OPM-2 cells treated with increasing dosage of rhDKK1 for 48 hours. (B) Representative protein level of CD138 in MM.1S and OPM-2 cells treated with rhDKK1 for 48 hours. (C) qPCR shows expressions of SDC1 of CD138+ plasma cells from 6 myeloma patients treated with rhDKK1 for 12 hours. (D) Representative protein levels of CD138 in MM cells treated with different dosage of rhDKK1 in presence of Wnt3a (200 ng/mL) or (E) WntC59 (100 nM) for 48 hours. (F) Schematic illustration of LEF1 binding sites, sequences, and positions on the SDC1 promoter. (G) Luciferase assay of a 1100-bp hSDC1-promoter in HEK293T cells transfected with increasing amount of TCF1 plasmid. (H) Western blotting shows CD138 protein level in MM.1S cells infected with increasing lentiviral carrying overexpression of LEF1 for 72 hr. (I) The luciferase assay of a 1100 bp hSDC1-promoter in HEK293T cells transfected with increasing amount of pCSV-TCF1 plasmid. (J) Representative western blotting showing the knockdown effects of short hairpin RNA (shRNA) targeting coding sequencing of LRP6 gene (shLRP6) compared with the NT-Ctrl in MM.1S and OPM-2 cells infected for 72 hours. (K) Representative of CD138 protein level in MM cells stably expressing shRNA targeting LRP6 or non-target control (NT Ctrl), and subsequently treated with rhDKK1 (100 ng/mL) for 48 hours. (L) Western blotting showing the cleavage of PARP to detect effect of rhDKK1, or (M) DKK1 neutralizing antibodies (1 μg/mL) on BTZ-induced apoptosis in MM cell with shLRP6 knockdown.
DKK1 maintains MM cell immatureness by downregulating CD138. (A) Quantitative PCR (qPCR) shows expressions of SDC1 (CD138) of MM.1S and OPM-2 cells treated with increasing dosage of rhDKK1 for 48 hours. (B) Representative protein level of CD138 in MM.1S and OPM-2 cells treated with rhDKK1 for 48 hours. (C) qPCR shows expressions of SDC1 of CD138+ plasma cells from 6 myeloma patients treated with rhDKK1 for 12 hours. (D) Representative protein levels of CD138 in MM cells treated with different dosage of rhDKK1 in presence of Wnt3a (200 ng/mL) or (E) WntC59 (100 nM) for 48 hours. (F) Schematic illustration of LEF1 binding sites, sequences, and positions on the SDC1 promoter. (G) Luciferase assay of a 1100-bp hSDC1-promoter in HEK293T cells transfected with increasing amount of TCF1 plasmid. (H) Western blotting shows CD138 protein level in MM.1S cells infected with increasing lentiviral carrying overexpression of LEF1 for 72 hr. (I) The luciferase assay of a 1100 bp hSDC1-promoter in HEK293T cells transfected with increasing amount of pCSV-TCF1 plasmid. (J) Representative western blotting showing the knockdown effects of short hairpin RNA (shRNA) targeting coding sequencing of LRP6 gene (shLRP6) compared with the NT-Ctrl in MM.1S and OPM-2 cells infected for 72 hours. (K) Representative of CD138 protein level in MM cells stably expressing shRNA targeting LRP6 or non-target control (NT Ctrl), and subsequently treated with rhDKK1 (100 ng/mL) for 48 hours. (L) Western blotting showing the cleavage of PARP to detect effect of rhDKK1, or (M) DKK1 neutralizing antibodies (1 μg/mL) on BTZ-induced apoptosis in MM cell with shLRP6 knockdown.
Because DKK1 is an antagonist of canonical Wnt signaling, we hypothesized the inhibitory effect of DKK1 on CD138 expression was through Wnt signaling. As speculated, a Wnt signaling activator, Wnt3a, successfully reversed the inhibitory effect of DKK1 on CD138 expression in MM.1S cells (Figure 2D). Similar to DKK1, the porcupine O-acyltransferase inhibitors, WntC59, also reduced CD138 expression in MM cells (Figure 2E). Promoter analysis implicates a consensus LEF-binding motif (Wnt-responsive element) locates upstream of the ATG translation start site in the CD138 promoter, whereas a consensus TCF-binding motif does not (Figure 2F). After ascending overexpression of LEF1, a gradient-dependent increased of transcriptional activity of CD138 promoter was observed (Figure 2G), accompanied with dose-dependent manner of CD138 augmentation (Figure 2H). By contrast, overexpression of transcriptional factor TCF did not evoke significant transcriptional activity of CD138 promoter (Figure 2I).
Because DKK1 competitively binds receptor LRP5/6 to antagonize canonical Wnt signaling,23 we established MM cells with LRP6 stably abolished and then treated with DKK1 (Figure 2J). DKK1 administration barely inhibited CD138 expression in MM cells with LRP6 knock-down (Figure 2K). In contrast, administration of DKK1 also had protective effects against BTZ-induced apoptosis in MM cells with LRP6 knockdown (Figure 2L; supplemental Figure 3A). Similarly, MM cells regulated with LRP6 knock-down had no influence on killing effect of DKK1 neutralizing antibodies (Figure 2M; supplemental Figure 3B). Taken together, these data suggested that excessive DKK1 in myeloma cells suppresses CD138 expression through canonical Wnt/β-catenin signaling pathway.
DKK1 induces drug resistance via CKAP4
Updated studied have identified that the cell surface receptor CKAP4 is a novel DKK1 receptor, and it stimulates signaling through phosphatidylinositide 3-kinase.14,15 We assumed that CKAP4 receptor may be a candidate transducer of DKK1 signaling in MM cells. Indeed, we found that the expression of CKAP4 was elevated in BR MM cells (Figure 3A), and augmented expressions of CKAP4 were also observed in MM patients suffering disease progression after BTZ-based treatments (Figure 3B). Notably, CKAP4 knockdown limited the protective effects of DKK1 against BTZ-induced apoptosis and weakened the anti-MM effect of DKK1 neutralizing antibodies (Figure 3C-D, F-G; supplemental Figure 3E-F). Nevertheless, MM.1S and OPM-2 cells with CKAP4 knockdown exhibited similar proliferation compared with their parental controls, but DKK1 administration still inhibited CD138 expression in CKAP4 knock-down MM cells (Figure 3E; supplemental Figure 3C-D). To ascertain DKK1 exerts protective effect against BTZ-induced apoptosis through CKAP4 receptor, we deleted the cysteine-rich domains CRD1 in DKK1 gene (Figure 3H-I), which is responsible for the interaction with CKAP4,14 and purified the exogenous proteins for MM treatment. Administration of a CRD1-defective DKK1 barely alleviated the cleavage of PARP compared with WT DKK1 (Figure 3J), and loss of protective effect on BTZ-induced apoptosis in MM cells (Figure 3K). Therefore, we speculate that DKK1 mainly induces drug resistance in MM cells via CKAP4 receptor.
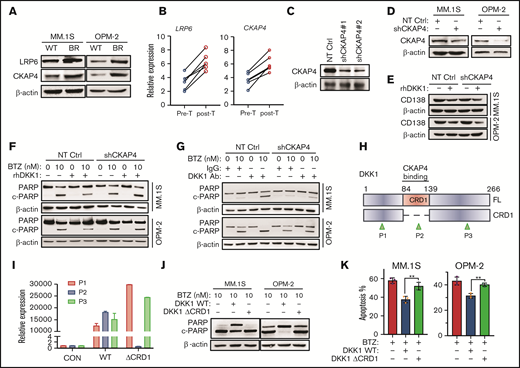
DKK1 induces drug resistance via receptor CKAP4. (A) Representative protein levels of LRP6 and CKAP4 in WT and BR MM cells. (B) LRP6 and CKAP4 expression in 6 patients with disease progression before and after BTZ-based treatment. (C) Western blotting shows the efficacy of 2 shRNAs targeting CKAP4 in HEK293T cells, and validates the knockdown efficacy of shRNA#1 in MM cells (D). (E) Representative of CD138 protein level in MM cells stably expressing shRNA targeting CKAP4 or non-target control (NT Ctrl), and subsequently treated with rhDKK1 (100 ng/mL) for 48 hours. (F) Western blotting shows the cleavage of PARP as a marker of apoptosis in MM cells stably expressing NT Ctrl or CKAP4-shRNA (CKAP4 KD) treated with BTZ and rhDKK1, or anti-DKK1 (G) for 24 hours. (H) Schematic illustration of CRD1 domain deletion in human DKK1 gene, and positions of 3 primers to validate the expression of truncations. CRD1, cysteine-rich domain; FL, full length; P1, position for primer 1; P2, position for primer 2; P3, position for primer 3 for the real-time PCR. (I) qPCR data shows DKK1 FL or ΔCRD1 truncation expressions using 3 different primers. (J) Cleavage of PARP in MM.1S and OPM-2 cells treated with FL or ΔCRD1 DKK1 in presence of BTZ for 24 hours. (K) Quantification of apoptotic MM.1S and OPM-2 cells treated with BTZ in presence of FL DKK1 or ΔCRD1 DKK1.
DKK1 induces drug resistance via receptor CKAP4. (A) Representative protein levels of LRP6 and CKAP4 in WT and BR MM cells. (B) LRP6 and CKAP4 expression in 6 patients with disease progression before and after BTZ-based treatment. (C) Western blotting shows the efficacy of 2 shRNAs targeting CKAP4 in HEK293T cells, and validates the knockdown efficacy of shRNA#1 in MM cells (D). (E) Representative of CD138 protein level in MM cells stably expressing shRNA targeting CKAP4 or non-target control (NT Ctrl), and subsequently treated with rhDKK1 (100 ng/mL) for 48 hours. (F) Western blotting shows the cleavage of PARP as a marker of apoptosis in MM cells stably expressing NT Ctrl or CKAP4-shRNA (CKAP4 KD) treated with BTZ and rhDKK1, or anti-DKK1 (G) for 24 hours. (H) Schematic illustration of CRD1 domain deletion in human DKK1 gene, and positions of 3 primers to validate the expression of truncations. CRD1, cysteine-rich domain; FL, full length; P1, position for primer 1; P2, position for primer 2; P3, position for primer 3 for the real-time PCR. (I) qPCR data shows DKK1 FL or ΔCRD1 truncation expressions using 3 different primers. (J) Cleavage of PARP in MM.1S and OPM-2 cells treated with FL or ΔCRD1 DKK1 in presence of BTZ for 24 hours. (K) Quantification of apoptotic MM.1S and OPM-2 cells treated with BTZ in presence of FL DKK1 or ΔCRD1 DKK1.
DKK1 activates NF-κB pathway via CKAP4
To determine which signaling pathway is affected by DKK1, we applied a screening experiment using the luciferase reporter system to monitor the activities of 17 different transcriptional factors. Our data indicated that activity of NF-κB reporter was the highest enhanced one among all transcriptional factors after DKK1 treatment, compared with the mildly activated E2F, MYC, and STAT3 (Figure 4A). To further identify the activation of these pathways, we assessed the phosphorylation of p65 and STAT3 in MM cells treated with DKK1, and western blotting results confirmed the p-p65 and p-STAT3 level increased in a dose-dependent manner, accompanying with suppression of IκB-α (Figure 4B). In addition, immunofluorescence staining and subcellular fractionation assay validated a nuclear accumulation of p65 after DKK1 administration (Figure 4C-D), and expressions of the downstream genes of NF-κB pathway were upregulated in a dose-dependent manner (Figure 4E). On the contrary, neither DKK1 lacking of CRD1 domain, nor CKAP4 knockdown on MM cells, could effectively activate NF-κB signaling thorough phosphorylating p65 and IκB-α (Figure 4F-G). Clinically, patient tissue with high level of DKK1 generally showed high percentage of positive p65 rate in bone marrow samples from relapsed patients (Figure 4H), and statistical analysis found a significant positive correlation between p65 and DKK1+ levels (Figure 4I). Thus, we further speculate that CKAP4 is a crucial factor to mediate DKK1 signal and evoke NF-κB pathway.
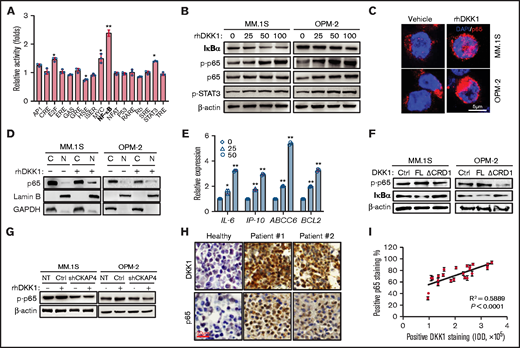
DKK1 activates NF-KB signaling pathway by CKAP4 receptor in multiple myeloma. (A) Fold changes in activations of 17 signaling pathways treated with PBS or rhDKK1 (100 ng/mL) for 24 hours in MM.1S cells. (B) Phosphorylation levels of p65 and STAT3, total IκB-α in MM.1S and OPM-2 cells treated with increasing dosage of rhDKK1 (0-100 ng/mL) for 1 hour. (C) Immunofluorescence staining to detect the nuclear localization of p65 in MM.1S and OPM-2 cells treated with PBS or rhDKK1 (100 ng/mL) for 12 hours. Scale bar, 5 μm. (D) Cytoplasmic and nuclear fraction of p65 in MM.1S and OPM-2 cells treated with PBS or rhDKK1 (100 ng/mL) for 12 hours. (E) qPCR detecting downstream target genes of NF-κB signaling under the stimulation of rhDKK1 (100 ng/mL) for 12 hours. (F) Levels of p-p65 and IκB-α protein in MM.1S and OPM-2 cells treated with full-length (FL) rhDKK1 or CRD1 domain depletion DKK1 (ΔCRD1) for 12 hours. (G) Levels of p-p65 and IκB-α protein in MM.1S and OPM-2 cells with CKAP4 receptor knockdown and treated with rhDKK1 (100 ng/mL) for 12 hours. (H) Immunohistochemistry staining of DKK1 and p65 proteins in the sequential slides of bone marrow biopsies from 2 patients and healthy donor. Scale bar, 20 μm. (I) Correlation coefficient between positive rate of p65 and DKK1 intensity in bone marrow tissues from MM patients (n = 12).
DKK1 activates NF-KB signaling pathway by CKAP4 receptor in multiple myeloma. (A) Fold changes in activations of 17 signaling pathways treated with PBS or rhDKK1 (100 ng/mL) for 24 hours in MM.1S cells. (B) Phosphorylation levels of p65 and STAT3, total IκB-α in MM.1S and OPM-2 cells treated with increasing dosage of rhDKK1 (0-100 ng/mL) for 1 hour. (C) Immunofluorescence staining to detect the nuclear localization of p65 in MM.1S and OPM-2 cells treated with PBS or rhDKK1 (100 ng/mL) for 12 hours. Scale bar, 5 μm. (D) Cytoplasmic and nuclear fraction of p65 in MM.1S and OPM-2 cells treated with PBS or rhDKK1 (100 ng/mL) for 12 hours. (E) qPCR detecting downstream target genes of NF-κB signaling under the stimulation of rhDKK1 (100 ng/mL) for 12 hours. (F) Levels of p-p65 and IκB-α protein in MM.1S and OPM-2 cells treated with full-length (FL) rhDKK1 or CRD1 domain depletion DKK1 (ΔCRD1) for 12 hours. (G) Levels of p-p65 and IκB-α protein in MM.1S and OPM-2 cells with CKAP4 receptor knockdown and treated with rhDKK1 (100 ng/mL) for 12 hours. (H) Immunohistochemistry staining of DKK1 and p65 proteins in the sequential slides of bone marrow biopsies from 2 patients and healthy donor. Scale bar, 20 μm. (I) Correlation coefficient between positive rate of p65 and DKK1 intensity in bone marrow tissues from MM patients (n = 12).
Because a CKAP4 mutant lacking the intracellular domain (ICD) also blocked NF-κB activation induced by exogenous DKK1 (supplemental Figure 3A; Figure 5A), we subsequently screen potential cytoplasmic proteins interacting with CKAP4. We ectopically expressed CKAP4-flag protein in HEK293T cells for mass spectrum assay after immunoprecipitation, and identified cullin associated and neddylation dissociated 1 (CAND1) as one of the components of CKAP4 complex (Figure 5B; supplemental Figure 3B). Furthermore, we immunoprecipitated CAND1-flag or CKAP4-flag fusion protein in HEK293T cells, and confirmed their tight interaction from both sides (Figure 5C). In addition, the interaction between endogenous CKAP4 and CAND1 could also be detected in MM.1S cells (Figure 5D). We found the interaction between CAND1 and CKAP4 increased significantly in presence of DKK1, and the binding of an E3 ligase cullin 1 (CUL1) was obviously attenuated on the contrary (supplemental Figure 4C; Figure 5E). Immunofluorescence also confirmed that CAND1 was recruited to the cell surface and colocalized with CKAP4 on the membrane (Figure 5F). To further determine the function of CAND1 in the CKAP4-mediated signal transduction, we established MM cells stably eliminating CAND1 (supplemental Figure 3D). CAND1 inhibition accelerated DKK1-induced NF-κB activation, as demonstrated by decreased expression of IκB-α and an enhancement in p-p65 (Figure 5G). At the same time, CAND1 knockdown effectively promoted ubiquitination of IκB-α and urged its instability (Figure 5H). When MM cells were treated with cycloheximide to terminate protein synthesis, the degradation of IκB-α protein was remarkably accelerated in MM cells with CAND1 depletion (Figure 5I). Because DKK1 competitively binds receptor CKAP4 to recruit CAND1 and then activates NF-κB signaling, we administrated DKK1 and anti-DKK1 in MM cells with CAND1 stably abolished. Interestingly, CAND1 is a downstream protein of the DKK1-CKAP4 axis, whether or not DKK1 or anti-DKK1 was added, thus CAND1 knock-downregulation had protective effects against BTZ-induced apoptosis in MM cells (Figure 5J-K; supplemental Figure 4E-F). Overall, our data suggest that DKK1 promotes the binding of CKAP4 to CAND1, thereby promoting the degradation of IκB-α by recruiting CAND1 to reduce the inhibition effect of CAND1 on the assembly of E3 ligase complex.
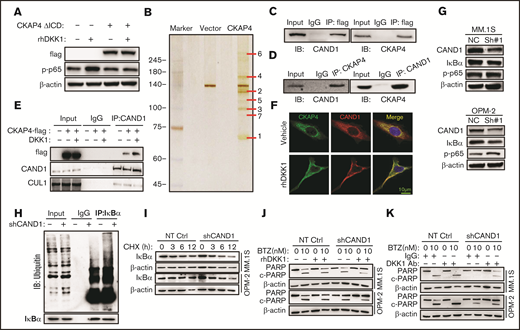
DKK1-CKAP4 axis activates NF-κB pathway thorough recruiting CAND1. (A) Levels of p-p65 and IκB-α in MM.1S cells bearing an ICD domain depletion CKAP4 (ΔICD) and treated with rhDKK1(100 ng/mL) for 12 hours. (B) Silver staining of lysate from HEK-293T cells transfected with vector or CKAP4-flag for 48 hours. (C) Coimmunoprecipitation (co-IP) assay shows interactions between exogenous CAND1-flag with CKAP4 or exogenous CKAP4-flag with CAND1 in HEK-293T cells. Input, 2% lysate. IP, M2-flag antibody. (D) Co-IP assay shows bilateral interactions between endogenous CAND1 and CKAP4 in MM.1S cells. (E) Alteration of interaction between CAND1 and CUL1 in HEK-293T cells transfected with vector control or CKAP4-flag for 48 hr and then treated with rhDKK1(100 ng/mL) for 12 hours. Input, 2% whole cell lysate. IP, anti-CUL1 antibody. (F) Immunofluorescence assay showing colocalization of CKAP4 with CAND1 in A549 cells treated with rhDKK1 (100 ng/mL) for 12 hours. Scale bar, 10 μm. (G) Levels of p-p65 and IκB-α in MM.1S and OPM-2 cells stably expressing NT Ctrl or CAND1-shRNA. (H) Ubiquitination status of IκB-α in HEK293T cells transfected with shCAND1 or NT Ctrl for 48 hours. (I) Degradation rate of IκB-α protein in MM.1S and OPM-2 cells with CAND1 knockdown and then treated with 20 μM cycloheximide for up to 12 hours. (J) Western blotting shows the cleavage of PARP as a marker of apoptosis in MM cells stably expressing NT Ctrl or CAND1-shRNA (CAND1 KD) treated with BTZ and rhDKK1, or (K) anti-DKK1 for 24 hours.
DKK1-CKAP4 axis activates NF-κB pathway thorough recruiting CAND1. (A) Levels of p-p65 and IκB-α in MM.1S cells bearing an ICD domain depletion CKAP4 (ΔICD) and treated with rhDKK1(100 ng/mL) for 12 hours. (B) Silver staining of lysate from HEK-293T cells transfected with vector or CKAP4-flag for 48 hours. (C) Coimmunoprecipitation (co-IP) assay shows interactions between exogenous CAND1-flag with CKAP4 or exogenous CKAP4-flag with CAND1 in HEK-293T cells. Input, 2% lysate. IP, M2-flag antibody. (D) Co-IP assay shows bilateral interactions between endogenous CAND1 and CKAP4 in MM.1S cells. (E) Alteration of interaction between CAND1 and CUL1 in HEK-293T cells transfected with vector control or CKAP4-flag for 48 hr and then treated with rhDKK1(100 ng/mL) for 12 hours. Input, 2% whole cell lysate. IP, anti-CUL1 antibody. (F) Immunofluorescence assay showing colocalization of CKAP4 with CAND1 in A549 cells treated with rhDKK1 (100 ng/mL) for 12 hours. Scale bar, 10 μm. (G) Levels of p-p65 and IκB-α in MM.1S and OPM-2 cells stably expressing NT Ctrl or CAND1-shRNA. (H) Ubiquitination status of IκB-α in HEK293T cells transfected with shCAND1 or NT Ctrl for 48 hours. (I) Degradation rate of IκB-α protein in MM.1S and OPM-2 cells with CAND1 knockdown and then treated with 20 μM cycloheximide for up to 12 hours. (J) Western blotting shows the cleavage of PARP as a marker of apoptosis in MM cells stably expressing NT Ctrl or CAND1-shRNA (CAND1 KD) treated with BTZ and rhDKK1, or (K) anti-DKK1 for 24 hours.
CKAP4 is a downstream target gene of IL-6
Our results have shown that DKK1 can activate the NF-κB signaling by CKAP4; we next intend to investigate how CKAP4 is regulated in MM cells. When examining the expression of CKAP4 among 13 MM cell lines, we found that ANBL-6 cell, which is IL-6 dependent, had the highest CKAP4 expression (Figure 6A; supplemental Figure 4A). Therefore, we speculate CKAP4 expression may be correlated with IL-6 level. Indeed, exogenous IL-6 could enhance the mRNA expression and protein level of CKAP4 in MM cells in a dose-dependent manner (Figure 6B-C). Because IL-6 stimulation leads to the activation of JAK/STAT pathway, and promoter analysis indicated 7 putative STAT1 binding motif located in the CKAP4 promoter (Figure 6D), we created a luciferase reporter that carries 1060 bps of CKAP4 promoter. The transcriptional activity of CKAP4-luc reporter was greatly enhanced upon STAT1 overexpression, but not STAT3 (Figure 6E; supplemental Figure 4B). In addition, binding of STAT1 transcriptional factor on DNA fragments containing the 7 binding motifs were all confirmed by polymerase chain reaction (PCR) based on chromatin immunoprecipitation (ChIP-PCR) assay (Figure 6F). Furthermore, addition of IL-6 also augmented the activation of NF-κB signaling by DKK1 (Figure 6G). On the other hand, overexpression of CKAP4 receptor facilitated, but knockdown of CKAP4 obstructed, the activation of NF-κB signaling, respectively (Figure 6H; supplemental Figure 4C). The correlation between CKAP4 and IL-6 expression was clinically confirmed in bone marrow samples from MM patients (Figure 6I). Overall, these data show that CKAP4 expression in MM cells is regulated by IL-6.
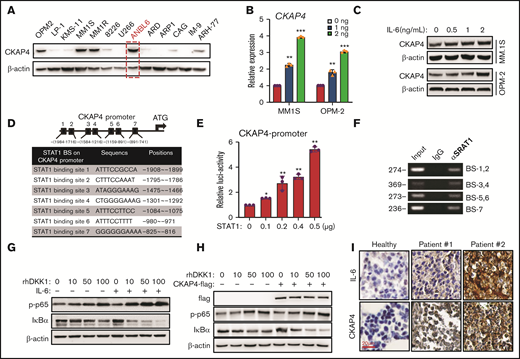
CKAP4 is under regulation of IL-6. (A) Western blotting shows CKAP4 levels in 13 myeloma cell lines. Red, IL-6–dependent cells. (B) CKAP4 mRNA expressions in MM.1S and OPM-2 cells treated with increasing dosage of IL-6 for 24 hours, and shows the protein levels (C). (D) Schematic illustration of STAT1 binding sites (BS), sequences and positions on the CKAP4 promoter. (E) Luciferase assay of a 1050-bp hCKAP4-promoter in HEK293T cells co-transfected with increasing amount of STAT1 plasmid. (F) The STAT1 BS on the CKAP4 promoter in MM.1S cells by ChIP-PCR. (G) Levels of p-p65 and IκB-α in MM.1S treated with increasing dosage of DKK1 with or without IL-6, or with or without CKAP4 overexpression (H) for 1 hour. (I) Immunohistochemistry staining of IL-6 and CKAP4 proteins in bone marrow biopsies from 2 patients and healthy donor. Scale bar, 100 μm.
CKAP4 is under regulation of IL-6. (A) Western blotting shows CKAP4 levels in 13 myeloma cell lines. Red, IL-6–dependent cells. (B) CKAP4 mRNA expressions in MM.1S and OPM-2 cells treated with increasing dosage of IL-6 for 24 hours, and shows the protein levels (C). (D) Schematic illustration of STAT1 binding sites (BS), sequences and positions on the CKAP4 promoter. (E) Luciferase assay of a 1050-bp hCKAP4-promoter in HEK293T cells co-transfected with increasing amount of STAT1 plasmid. (F) The STAT1 BS on the CKAP4 promoter in MM.1S cells by ChIP-PCR. (G) Levels of p-p65 and IκB-α in MM.1S treated with increasing dosage of DKK1 with or without IL-6, or with or without CKAP4 overexpression (H) for 1 hour. (I) Immunohistochemistry staining of IL-6 and CKAP4 proteins in bone marrow biopsies from 2 patients and healthy donor. Scale bar, 100 μm.
Targeting DKK1-CKAP4-IL-6 axis enhances sensitivity to bortezomib treatment of MM cells in vitro and in vivo
Our data have revealed that DKK1/IL-6-CKAP4 axis played an important role in drug resistance of MM cells. Eventually, we aim to clarify the significance of targeting the DKK1-IL-6/CKAPS4 axis in overcoming BTZ resistance in myeloma. When DKK1 and IL-6 neutralizing antibodies was present in BR-MM cells treated with BTZ or CFZ, significant higher apoptosis rates were elicited compared with the control (Figure 7A-B; supplemental Figure 6A). By comparison, DKK1 and IL-6 neutralizing antibodies did not alter the drug sensitivity of MM cells to melphalan or dexamethasone (supplemental Figure 6B-C). Collectively, these results argue that combination DKK1 and IL-6 neutralizing antibodies specifically enhance the resistance of myeloma cells to PIs. We established BR MM.1S cells with CKAP4 stably silenced, and injected these cells and counterpart control cells subcutaneously into the dorsal lateral flank of NSG mice. After 3 weeks, when the established tumor volume reached approximately 500cmm3, mice received intraperitoneal injections of PBS or BTZ (0.5 mg/kg, every 3 days). As expected, tumor growth was significantly suppressed only in mice bearing CKAP4-KD cells, but not the parental control cells (Figure 7C; supplemental Figure 6D), and the overall survival rate of mice bearing CKAP4 ablation MM cells was significantly improved (Figure 7D). Meanwhile, CKAP4 KD MM cells displayed higher sensitive to BTZ, as evidenced by increased account of terminal deoxynucleotidyltransferase-mediated dUTP nick end labeling-positive apoptotic cells (Figure 7E).
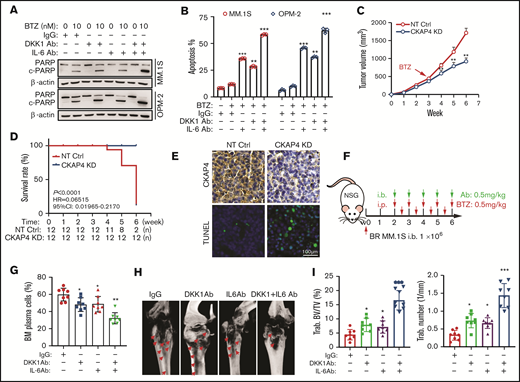
Targeting IL-6 and DKK1 overcomes BTZ-drug resistance in vivo. (A) Cleavage of PARP as the apoptotic marker in MM cells treated with or without anti-DKK1, anti-IL-6, combined with BTZ for 24 hours (n = 3). (B) Quantification of apoptotic MM cells treated with BTZ (0-10 nM) with or without anti-DKK1 or anti-IL-6 for 24 hours (n = 3). (C) Tumor growth of BTZ-resistant (BR) MM.1S cells stably expressing CKAP4-shRNA (CKAP4 KD) or NT Ctrl (3 × 106 cells/mouse, n = 12) in NSG mice receiving BTZ (0.5 mg/kg) at week 3 after inoculation for another 4 weeks. (D) Survival rate of mice at the time point of tumor diameter over 15 mm (n = 12). (E) Immunohistochemistry staining in tissues from xenograft of different mice groups to detect CKAP4 level and apoptosis using a terminal deoxynucleotidyltransferase-mediated dUTP nick end labeling kit. Scale bar, 100 μm. (F) Experiment design and treatment schedule in a mice intra-bone (i.b.) model harboring BR-MM.1S cells (n = 12). (G) Quantification of the percentage of CD138+ plasma cells in bone marrow biopsies using flow cytometry. (H) Representative micro-computed tomography images of mouse femurs bearing BR MM.1S cells and treated with IgG, anti-DKK1, and anti-IL-6 neutralizing antibodies (0.5 mg/kg) in presence of BTZ (0.5 mg/kg) (n = 8). (I) Measurement of the percentage of bone volume to total volume (BV/TV) and trabecular number in the metaphyseal regions of the mice femurs in the IgG, anti-DKK1, anti-IL-6 and anti-DKK1+ anti-IL-6 neutralizing antibodies groups were analyzed (n = 8).
Targeting IL-6 and DKK1 overcomes BTZ-drug resistance in vivo. (A) Cleavage of PARP as the apoptotic marker in MM cells treated with or without anti-DKK1, anti-IL-6, combined with BTZ for 24 hours (n = 3). (B) Quantification of apoptotic MM cells treated with BTZ (0-10 nM) with or without anti-DKK1 or anti-IL-6 for 24 hours (n = 3). (C) Tumor growth of BTZ-resistant (BR) MM.1S cells stably expressing CKAP4-shRNA (CKAP4 KD) or NT Ctrl (3 × 106 cells/mouse, n = 12) in NSG mice receiving BTZ (0.5 mg/kg) at week 3 after inoculation for another 4 weeks. (D) Survival rate of mice at the time point of tumor diameter over 15 mm (n = 12). (E) Immunohistochemistry staining in tissues from xenograft of different mice groups to detect CKAP4 level and apoptosis using a terminal deoxynucleotidyltransferase-mediated dUTP nick end labeling kit. Scale bar, 100 μm. (F) Experiment design and treatment schedule in a mice intra-bone (i.b.) model harboring BR-MM.1S cells (n = 12). (G) Quantification of the percentage of CD138+ plasma cells in bone marrow biopsies using flow cytometry. (H) Representative micro-computed tomography images of mouse femurs bearing BR MM.1S cells and treated with IgG, anti-DKK1, and anti-IL-6 neutralizing antibodies (0.5 mg/kg) in presence of BTZ (0.5 mg/kg) (n = 8). (I) Measurement of the percentage of bone volume to total volume (BV/TV) and trabecular number in the metaphyseal regions of the mice femurs in the IgG, anti-DKK1, anti-IL-6 and anti-DKK1+ anti-IL-6 neutralizing antibodies groups were analyzed (n = 8).
At the same time, BR MM cells were injected into the femur bone marrow of NSG mice, which then were treated with BTZ together with neutralizing antibodies of DKK1 and/or IL-6 after 2 weeks of inoculation. After being treated for 4 weeks, the bone marrow of mice was flushed and subjected to flow cytometry (Figure 7F). As shown, percentage of CD138+ cells in the bone marrow of NOD/SCID mice were greatly suppressed by the combination of neutralizing antibodies of DKK1 and IL-6 in presence of BTZ, but neither DKK1 nor IL-6 antibody solo administration significantly augmented the antitumor effect of BTZ (Figure 7G). Strikingly, the combination of neutralizing antibodies of DKK1 and IL-6 also effectively alleviated bone lesion of mice destroyed by MM cells (Figure 7H), as evidenced by higher percentages of bone volume to total volume and trabecula numbers at the metaphysis and diaphysis (Figure 7I). Obviously, manipulating the DKK1-IL-6 axis with both DKK1 and IL-6 neutralizing antibodies has a clearly additive anti-MM effect and attenuate bone destruction in vivo when used in combination with PIs.
Discussion
In the current study, we report a previously not known role of DKK1 in PI-induced drug resistance thorough canonical Wnt signaling and noncanonical NF-κB signaling pathways, respectively. Mechanistically, DKK1 drives CD138 downregulation to promote myeloma malignancy via inhibition of canonical Wnt signaling; on the other hand, DKK1 induces activation of NF-κB signaling via CKAP4 receptor. Additionally, IL-6 stimulates CKAP4 expression to generate drug resistance via a paracrine or autocrine manner. Our study also suggests that manipulating DKK1-IL-6/CAKP4 axis may enhances sensitivity to PI treatment, and sheds light on developing novel therapeutic strategies to overcome drug resistance and osteolytic lesions in MM patients.
DKK1, an antagonist of Wnt signaling, binds to the Wnt coreceptor LRP5/6, masks the active sites of LRP5/6, thereby triggers their endocytosis and makes it unavailable for interactions with Wnt ligands.24 Abundant level of DKK1 is a hallmark of malignant MM, and frequently found in the myeloma microenvironment of patients with serious bone disease, but highly restricted in normal tissues.25 Moreover, overexpression of DKK1 has been described as a negative prognostic factor.26,27 Previous study has presented evidence that DKK1 is responsive to c-Jun N-terminal kinase (JNK) signaling in MM plasma cells, and oxidative stress enhances DKK1 expression through the JNK cascade.28 Obviously, abnormal expression of DKK1 is decided by genetic context of MM cells and specific stimuli within the bone marrow microenvironment. Herein, our data showed that DKK1 expression was greatly enhanced in BTZ-resistant MM cells and in clinical patients treated with BTZ-based regimens. Seemingly, DKK1 level could be used as a hallmark for monitoring the sensitivity of myeloma cells and the appearance of induced chemoresistance.
Although accumulating evidence has established a definite link between DKK1 and myeloma bone disease, it remains obscure about the precise effects and mechanisms of DKK1 in the pathogenesis of MM. Yaccoby et al found that antibody-based inhibition of DKK1 in mice could mitigate MM growth and tumor-induced bone resorption in vivo.29 However, in vitro experiments excluded a direct cytotoxic effect of anti-DKK1 antibody on MM cell lines, because blocking DKK1 could only attenuate MM cells proliferation in the presence of BMSCs, thorough an IL-6–dependent manner.11 In this study, we found that DKK1 alone unaffected the growth of MM cells, but was sufficient to render drug resistance to PIs. Further investigation showed that DKK1 downregulated CD138 to maintain the tumor cell at immature status and promotes malignancy. CD138, also known as syndecan-1, is a heparin sulfate proteoglycan expressed at distinct differentiation stages of B lymphoid cells such as pre-B cells, immature B cells, and immunoglobulin-producing plasma cells.30 It is speculated that CD138 regulates adhesion and survival of MM cells in the bone marrow niche.31 However, CD138 expression is frequently downregulated from the plasma membrane when myeloma cells outgrow, allowing malignant cells to invade and metastasize. A study revealed that MM cells with low levels of CD138 displayed an immature phenotype and lower sensitivity to lenalidomide.23 Furthermore, CD138 downregulation has been observed during the course of clinical therapy in some MM patients.32 Therefore, a reduction in CD138 expression is associated with MM malignancy and poor prognosis. However, it remains unclear how CD138 downregulation happens in MM. Herein, our results indicated that BTZ-resistant myeloma cells produced excessive DKK1 to inhibit CD138 expression. Previous evidences have shown that CD138 can bind with Wnt ligands and modulate paracrine Wnt pathway activation. Our data showed that Wnt3a could enhance, whereas DKK1 could inhibit CD138 expression, probably through LRP6-mediated canonical Wnt signaling, because when LRP6 expression was interfered, neither the anti-apoptosis effect of DKK1, nor pro-apoptosis of DKK1 neutralizing antibody could be elicited. Indeed, there exists consensus TCF/LEF-binding motif (Wnt-responsive element) located upstream of CD138 promoter. Therefore, our data suggested that excessive DKK1 from BTZ-resistant cells can drive CD138 downregulation via antagonism of Wnt signal.
In this study, we found that DKK1 mainly induces drug resistance in MM cells via CKAP4 receptor. DKK1 interacts with CKAP4 and LRP6 with similar affinity but binds with these 2 receptors via 2 different cysteine-rich domains, CRD1 and CRD2, respectively.15 Previous study has revealed that CKAP4 can activate AKT by forming a complex with phosphatidylinositide 3-kinase upon the binding of DKK1, thereby leading to enhanced growth of tumor cells.16 In the present study, we observed that DKK1 evoked the activation of NF-κB pathway to enhance drug resistance via CKAP4. NF-κB pathway is a crucial regulator of drug resistance and frequently elevated in BTZ-resistant primary MM cells from patients.33,34 We found DKK1 exposure elevated the phosphorylation levels and nuclear accumulation of NF-κB subunits p65, thorough a rapid phosphorylation and degradation of IκB-α. Mechanistically, binding of DKK1 on CKAP4 receptor recruits CAND1 from the cullin 1 E3 ligase complex, consequentially release the ubiquitination and degradation of IκB-α. Herein, our data also showed that DKK1 could enhance drug resistance of MM cells via autocrine or paracrine IL-6. IL-6 directly increased CKAP4 expression, and JAK/STAT pathway was required in this process; thus, IL-6 secreted from BMSCs or MM cells can regulate CKAP4 expression and then facilitate the generation of drug resistance.
Overall, our results implicate that the DKK1-CKAP4 axis is a crucial regulator for PI resistance in myeloma via stimulating NF-κB pathway. The significance of our study is to develop an indicator for chemotherapeutic response, and develop combination treatment strategies targeting DKK1-IL-6 axis to overcome myeloma relapse eventually. However, 1 limitation of our study is that most of our combination treatment evaluations were carried out in BTZ-resistant MM cells but not their counterpart controls, although both interventions resulted in obvious improvement in tumor burden, whether these combinations are suitable for WT MM cells, or newly diagnosed MM patients in clinic should be considered.
Acknowledgments
The authors thank Kai Zhang at the Department of Biochemistry and Molecular Biology, Tianjin Medical University, for mass spectrum assays.
This work was supported by the National Natural Science Foundation of China (81870161, 82070221, Z.L.; 81900215, J.Y. Wang), the Beijing Natural Science Foundation of China (Z200020), the Talent Project of Tianjin Medical University (11601501/2016KJ0317, Z.L.), the College Student Innovation Program of Tianjin Medical University (202010062030, Z.Y.P.), and the Tianjin Research Innovation Project for Postgraduate Students (2019YJSB110, JJ.W.; 2020YJSB162, Y.X.).
Authorship
Contribution: X.L. and Z.Q.L. contributed to writing the manuscript; J.J.W., S.Z., J.X.Z., Y.X., H.M.J., J.G., Y.X.W., Z.Y.P., and M.Q.W. contributed to performing the experiments and statistical analyses; X.L., Y.X., and S.W. were in charge of the animal studies; Y.P.Z. provided the patient samples and clinical statistics; and Z.L. and Y.P.Z. contributed to the design of the experiments.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
For data sharing, contact the corresponding author: zhiqiangliu@tmu.edu.cn.
Correspondence: Yuping Zhong, Department of Hematology, Qingdao Municipal Hospital, School of Medicine, Qingdao University, Qingao, Shandong 266021, China; e-mail: zhongyp3352@126.com; or Zhiqiang Liu, Department of Physiology and Pathophysiology, School of Basic Medical Science, Tianjin Medical University, Heping, Tianjin 300070, China; e-mail: zhiqiangliu@tmu.edu.cn.

