

Key Points
We generated cell lines that overexpress wild-type or mutant BTK (BTKC481S and BTKC481R) and mimic ibrutinib-sensitive and -resistant CLL.
We developed xenograft mouse models by transplanting these cells into Rag2−/−γc−/− mice to characterize in vivo disease.

Abstract
Although ibrutinib improves the overall survival of patients with chronic lymphocytic leukemia (CLL), some patients still develop resistance, most commonly through point mutations affecting cysteine residue 481 (C481) in Bruton’s tyrosine kinase (BTKC481S and BTKC481R). To enhance our understanding of the biological impact of these mutations, we established cell lines that overexpress wild-type or mutant BTK in in vitro and in vivo models that mimic ibrutinib-sensitive and -resistant CLL. MEC-1 cell lines stably overexpressing wild-type or mutant BTK were generated. All cell lines coexpressed GFP, were CD19+ and CD23+, and overexpressed BTK. Overexpression of wild-type or mutant BTK resulted in increased signaling, as evidenced by the induction of p-BTK, p-PLCγ2, and p-extracellular signal–related kinase (ERK) levels, the latter further augmented upon IgM stimulation. In all cell lines, cell cycle profiles and levels of BTK expression were similar, but the RNA sequencing and reverse-phase protein array results revealed that the molecular transcript and protein profiles were distinct. To mimic aggressive CLL, we created xenograft mouse models by transplanting the generated cell lines into Rag2−/−γc−/− mice. Spleens, livers, bone marrow, and peripheral blood were collected. All mice developed CLL-like disease with systemic involvement (engraftment efficiency, 100%). We observed splenomegaly, accumulation of leukemic cells in the spleen and liver, and macroscopically evident necrosis. CD19+ cells accumulated in the spleen, bone marrow, and peripheral blood. The overall survival duration was slightly lower in mice expressing mutant BTK. Our cell lines and murine models mimicking ibrutinib-resistant CLL will serve as powerful tools to test reversible BTK inhibitors and novel, non–BTK-targeted therapeutics.
Introduction
The B-cell receptor (BCR) pathway is responsible for proliferation, maintenance, and survival of normal and malignant B cells,1 including those in chronic lymphocytic leukemia (CLL). A pivotal enzyme in the BCR axis is Bruton’s tyrosine kinase (BTK),2 which signals to phospholipase C-γ2 (PLCγ2) and multiple signaling cascades, leading to changes in cell metabolism, transcription, and translation.3-5 The importance of the BCR pathway, the pathophysiology of CLL, and the prominence of BTK in the BCR signalosome suggest that BTK may be a good therapeutic target.
Ibrutinib binds covalently to the cysteine 481 (C481) residue of the kinase and irreversibly inactivates BTK.6 Inactivation of BTK results in inhibition of proliferation and dissociates B cells from microenvironmental signals in the lymph nodes, leading to their migration to the peripheral blood.7
Preclinical studies using primary CLL cells and TCL-1 mouse models have suggested that ibrutinib is efficacious in several B-cell malignancies.5,8,9 In the clinic, ibrutinib resulted in impressive overall and progression-free survival with very low untoward toxicity. Thus, the US Food and Drug Administration approved ibrutinib for the treatment of patients with previously treated CLL10 and for elderly patients with CLL,11 disease with poor prognosis (eg, those with 17p deletions),12 and treatment-naive patients.13
Although at the early stage, a small percentage of patients develop CLL that is resistant to ibrutinib,14 the number steadily increases as the length of follow-up increases. Some patients have Richter’s transformation, whereas many show CLL progression. Several studies have identified mutations in 2 enzymes in the BCR pathway in prerelapse and postrelapse disease during progression of CLL in patients receiving ibrutinib therapy. These include BTK and its immediate downstream kinase, PLCG2.15-18 Of these mutations, the most common are point mutations in the C481 binding site in BTK. Such mutations prevent irreversible drug binding on the C481 site, conferring ibrutinib resistance. For the BTK C481 residue, the site of ibrutinib binding, cysteine-to-serine (C481S) and cysteine-to-arginine (C481R) mutations are the most prevalent.14 These same mutations are expected to occur with the use of second-generation BTK inhibitors, such as acalabrutinib and zanubrutinib.19-21
These data underscore the need to better understand the biology of BTK-mutated CLL and develop new agents that are effective against BTK C481 mutations. However, it is challenging to understand the biology of BTK C481 mutations, because there are currently no models for both in vitro and in vivo use that mimic C481-mutation–related, ibrutinib-resistant CLL. Such a model has been created for MYD88-mutated Waldenström macroglobulinemia and activated B-cell–type diffuse large B-cell lymphoma cells.22 In CLL, a patient-derived xenograft model has been tested.23
In the current study, we developed ibrutinib-resistant CLL cell lines harboring BTK C481S and C481R mutations, compared the biology of these mutant cell lines to that of cell lines with wild-type (WT) BTK, and developed and characterized mouse models with WT and mutant BTK cell lines. Those models could be extended to other B-cell diseases treated with ibrutinib, such as mantle cell lymphoma and Waldenström macroglobulinemia.
Materials and methods
Cell lines and cell cultures
We selected the MEC-1 cell line for this study because it was established using the peripheral blood of a patient with B-cell CLL in prolymphocytoid transformation, and a reproducible xenograft murine CLL model had been developed using this cell line.24,25 A chromosome analysis of this cell line has been published.24 Details of the generation of cell lines are provided in the supplemental Methods. All cell lines were routinely screened for Mycoplasma species using a MycoAlert Mycoplasma Detection Kit (Lonza). Parent cell lines were authenticated by the Cytogenetics and Cell Authentication Core Facility at MD Anderson Cancer Center.
RNA-sequencing assay
Total RNA was isolated from cells by using an RNeasy Mini Kit (Qiagen) according to the manufacturer’s instructions. Samples were sequenced by the Sequencing and Noncoding RNA Program at MD Anderson Cancer Center. Details are provided in the supplemental Methods.
Reverse-phase protein array assay
Exponentially growing MEC-1 cells were seeded (1 × 107 per group) and incubated for 24 hours. The cells were lysed with 1× lysis buffer, and the cellular proteins were denatured in 1% sodium dodecyl sulfate containing β-mercaptoethanol. Lysates were serially diluted and printed on nitrocellulose-coated plates (Grace Bio-Labs) at the Functional Proteomics Reverse Phase Protein Array (RPPA) Core Facility at MD Anderson. Details are provided in the supplemental Methods.
In vivo experiments
We used modified MEC-1 cells to establish a xenograft mouse model.25 Mice were randomly assigned to 4 groups: no disease (n = 8), BTKWT (n = 10), BTKC481S (n = 10), and BTKC481R (n = 10). Specifically, 1 × 107 MEC-1 cells (BTKWT, BTKC481S, and BTKC481R) were injected IV into 8-week-old female Rag2−/−γc−/− mice that were monitored daily for development of leukemia and progression. Individual mice were euthanized when they became moribund. At this point, the spleen, liver, left femur, and peripheral blood samples were collected. Cells were isolated and stained with a monoclonal antibody against human CD19-phycoerytherin (PE) clone J3119 (Beckman Coulter), and flow cytometry analysis was performed.
The Institutional Animal Care and Use Committee approved and supervised all animal studies. The mice were cared for in accordance with the guidelines set forth by the American Association for Accreditation of Laboratory Animal Care and the Guide for the Care and Use of Laboratory Animals issued by the US Public Health Service.
Statistical analysis
Unless specified otherwise, all data are expressed as the mean ± SD of results from at least 3 independent experiments. Two-sided Student t tests were used to test the relationships between group means. Survival analysis, Student t tests, and analyses of variance were calculated with GraphPad Prism software. P-values indicate the probability that the difference in the means was due to chance. Other statistical analyses were performed in R software. P < .05 denoted statistically significant results.
Results
In vitro characterization of WT and mutant-BTK–expressing cells
We transduced MEC-1 cells to generate cell lines that stably overexpressed GFP, BTKWT, and the 2 BTK variants BTKC481S and BTKC481R. Overexpression of WT or mutant BTK resulted in increased signaling, as evidenced by levels of induced p-BTK, p-PLCγ2, and phospho-extracellular signal-regulated kinase (p-ERK; Figure 1A). Phospho- and total-BTK levels increased in cells that overexpressed BTKWT and BTKC481R, whereas p-ERK levels increased in all BTK-overexpressing cells upon IgM stimulation (Figure 1B). Ibrutinib treatment dramatically decreased the p-BTK and pERK levels in BTKWT cells. In BTKC481S, pBTK levels did not change at lower concentrations (0.01 and 0.1 µM); however, the levels decreased at the highest concentration (1 µM). In addition, pERK levels did not change in BTKC481S and slightly decreased (10% to 15% inhibition) in BTKC481R upon incubation with ibrutinib for 3 hours (Figure 1C).
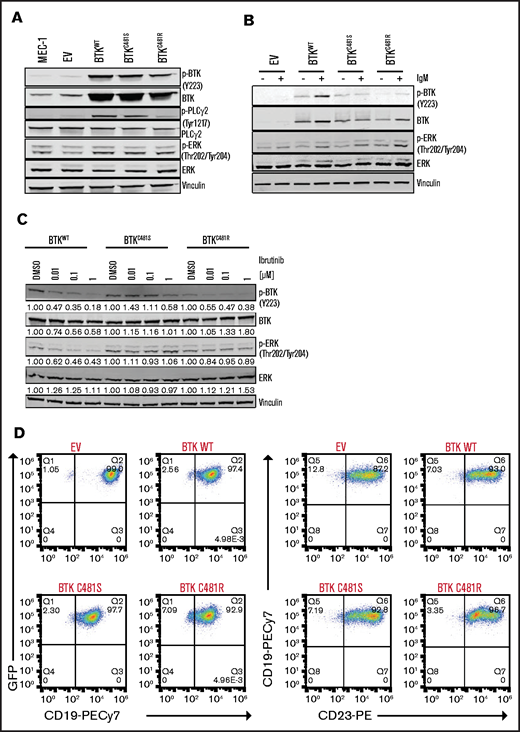

In vitro characterization of MEC-1 cells overexpressing WT and mutant BTK. GFP-labeled MEC-1 cell lines stably overexpressing mutant BTK (BTKC481S or BTKC481R) and BTKWT were generated by using standard lentiviral transduction methods. Cells were sorted to enrich the transduced GFP+ cell populations in each cell line. For all experiments, a population of cells >75% GFP+ was used. (A) Phosphorylated (Y223) and total BTK proteins were overexpressed in transduced cells. p-PLCγ2 (Tyr1217) and p-ERK (Thr202/Tyr204) levels were also increased in all BTK-overexpressing cells. Vinculin was the loading control. (B) IgM stimulation in transduced MEC-1 cells. Cells (5 ×105 per milliliter) were seeded in flasks, with or without IgM (20 µg/mL) and incubated at 37°C for 15 minutes. Protein extracts were subjected to immunoblot assays to determine the levels of pBTK (Y223), BTK, pERK (Thr202/Tyr204), and ERK. Vinculin was the loading control. (C) Exponentially growing GFP+ cells were seeded in flasks and incubated with ibrutinib (0.01, 0.1, and 1 µM) for 3 hours. Protein extracts were subjected to immunoblot assays to determine the levels of pBTK (Y223), BTK, pERK (Thr202/Tyr204), and ERK. Vinculin was the loading control. (D) Validation of cell surface markers. Cells were stained with anti-CD19-PECy7 and anti-CD23-PE monoclonal antibodies and analyzed with a flow cytometer. Flow cytometry dot plots showing GFP+ cells (left) and CD19+CD23+ cells (right) after gating on the GFP+ cell population.
In vitro characterization of MEC-1 cells overexpressing WT and mutant BTK. GFP-labeled MEC-1 cell lines stably overexpressing mutant BTK (BTKC481S or BTKC481R) and BTKWT were generated by using standard lentiviral transduction methods. Cells were sorted to enrich the transduced GFP+ cell populations in each cell line. For all experiments, a population of cells >75% GFP+ was used. (A) Phosphorylated (Y223) and total BTK proteins were overexpressed in transduced cells. p-PLCγ2 (Tyr1217) and p-ERK (Thr202/Tyr204) levels were also increased in all BTK-overexpressing cells. Vinculin was the loading control. (B) IgM stimulation in transduced MEC-1 cells. Cells (5 ×105 per milliliter) were seeded in flasks, with or without IgM (20 µg/mL) and incubated at 37°C for 15 minutes. Protein extracts were subjected to immunoblot assays to determine the levels of pBTK (Y223), BTK, pERK (Thr202/Tyr204), and ERK. Vinculin was the loading control. (C) Exponentially growing GFP+ cells were seeded in flasks and incubated with ibrutinib (0.01, 0.1, and 1 µM) for 3 hours. Protein extracts were subjected to immunoblot assays to determine the levels of pBTK (Y223), BTK, pERK (Thr202/Tyr204), and ERK. Vinculin was the loading control. (D) Validation of cell surface markers. Cells were stained with anti-CD19-PECy7 and anti-CD23-PE monoclonal antibodies and analyzed with a flow cytometer. Flow cytometry dot plots showing GFP+ cells (left) and CD19+CD23+ cells (right) after gating on the GFP+ cell population.
CLL is defined as the expansion of monoclonal, mature CD5+CD23+ B cells in the peripheral blood, secondary lymphoid tissues, and bone marrow,26 and CLL cells typically coexpress CD19, CD5, and CD23. We determined that >90% of the cells in each of our cell lines showed GFP, CD19, and CD23 positivity (Figure 1D). Because all transduced cell lines coexpressed GFP, we were able to enrich the transfected population by using flow cytometry to sort GFP+ cells. Representative images captured by fluorescence microscopy also confirmed that GFP was expressed in all of the generated cell lines (Figure 1E).
The growth rates of the MEC-1 cell lines with WT and mutant BTK were similar; however, cells harboring BTKC481R showed slower proliferation rates, which was significantly different from the empty vector (EV), BTKWT, and BTKC481S cell lines (Figure 1F). Furthermore, we did not observe any distinct distributions of populations in cell cycle profiles, and the differences between group means among the cell lines were not statistically significant (Figure 1G). The percentages of cells in each cell cycle phase are presented in supplemental Table 2. Overall, these data demonstrate that all generated cell lines are CD19+ and CD23+, coexpress GFP, overexpress BTK, and have active BCR signaling and similar cell cycle profiles.
Transcriptomic profiling of WT and mutant-BTK-expressing cells
To identify potential molecular subtypes and associated pathway features in these cell lines, we performed RNA sequencing (RNAseq)–based transcriptomic profiling. The differentially expressed genes (DEGs) showed significant (greater than twofold) changes in transcriptional expression (q < .05). Principal component analysis showed a clear separation between BTKEV cells and the cell lines overexpressing BTK (BTKWT, BTKC481S, and BTKC481R; supplemental Figure 1). Overexpression of WT BTK led to 166 upregulated and 69 downregulated DEGs in BTKWT cells, compared with BTKEV cells (supplemental Figure 2A). The comparison of all BTK-overexpressing cell lines with BTKEV revealed 80 upregulated and 15 downregulated DEGs in common (supplemental Figure 2B-C). The overexpression of WT BTK (BTKWT vs BTKEV) led to DEGs that were involved in various signaling pathways, such as those for axonal guidance, G-protein–coupled receptor, ephrin receptor, cyclic adenosine 5′-monophosphate–mediated, paxillin, and CXCR4 signaling (supplemental Figure 3A). Among the DEGs identified, there were 19 transcription factors (AFF3, ETS1, FHL5, FOXO3, GLIS3, HLX, ID1, KLF9, KLF11, LEF1, LMO4, NFATC1, SIX4, TBX15, VDR, ZEB2, ZPF30, ZNF114, and ZNF532). The top 3 canonical pathways that were associated with these transcription factors were the epithelial-mesenchymal–transition pathway (ETS1, LEF1, and ZEB2), senescence pathway (ETS1, FOXO3, NFATC1), and interleukin-7 (IL-7) signaling pathway (FOXO3, NFATC1). In addition, there were 11 transmembrane receptors, 13 transporter proteins, 9 G-protein–coupled receptors, and 7 cytokines altered in BTKWT compared with BTKEV cells.
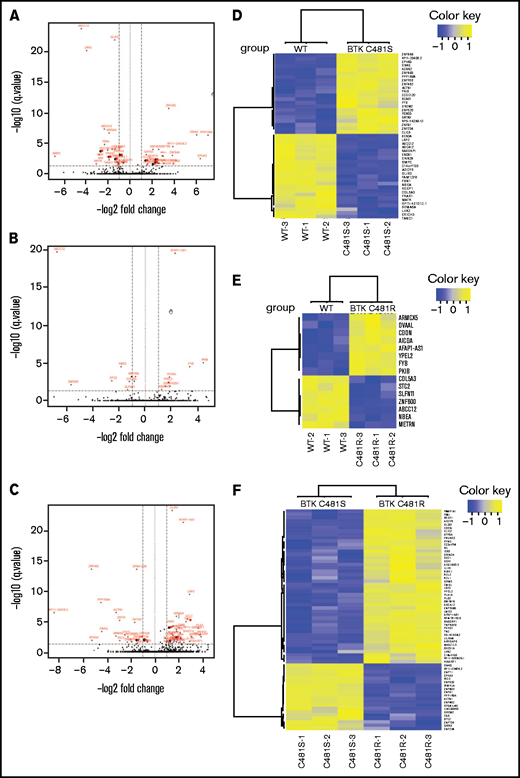
Comparison of the transcriptomic profiles of WT and mutant-BTK–overexpressing cells. (A-C) Volcano plots comparing MEC-1 cells overexpressing BTKWT and BTKC481S (A), BTKWT and BTKC481R (B), and BTKC481S and BTKC481R (C). The DEGs (FDR, ≤0.05; fold change, >2 or < −2) are labeled in red. (D-F) Heat maps of the DEGs (FDR, ≤0.05; fold change, >2 or <−2) between MEC-1 cells overexpressing BTKWT and BTKC481S (D), BTKWT and BTKC481R (E), or BTKC481S and BTKC481R (F).
Comparison of the transcriptomic profiles of WT and mutant-BTK–overexpressing cells. (A-C) Volcano plots comparing MEC-1 cells overexpressing BTKWT and BTKC481S (A), BTKWT and BTKC481R (B), and BTKC481S and BTKC481R (C). The DEGs (FDR, ≤0.05; fold change, >2 or < −2) are labeled in red. (D-F) Heat maps of the DEGs (FDR, ≤0.05; fold change, >2 or <−2) between MEC-1 cells overexpressing BTKWT and BTKC481S (D), BTKWT and BTKC481R (E), or BTKC481S and BTKC481R (F).
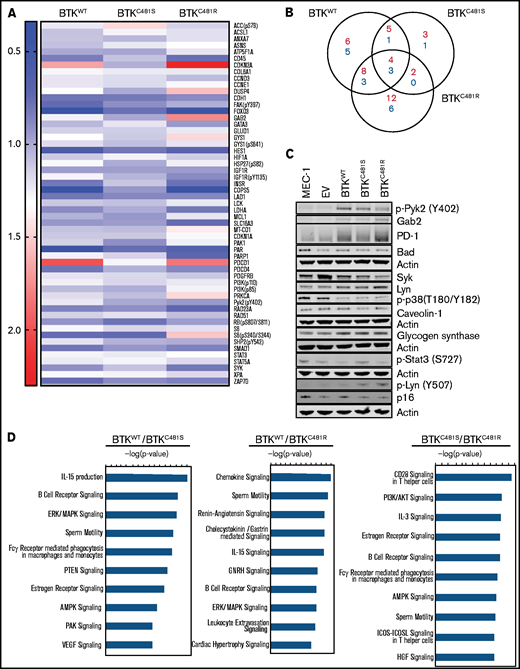
Functional protein profiling of WT and mutant-BTK–overexpressing MEC-1 cells. Protein was extracted in biological triplicates from exponentially growing MEC-1 cells with either WT or mutant BTK and subjected to RPPA assays that included 426 antibodies. (A) Representative heat map of proteins with increased and decreased expression in each cell line (n = 3 per cell line). All BTK-overexpressing cell lines (BTKWT, BTKC481S, and BTKC481R) were compared with the BTKEV cell line. The fold changes of mean linear values were used to generate the heat map (fold change, ≥1.2 or ≤0.8). The color key indicates the fold change. (B) The number of increased (red) and decreased (blue) phosphorylated and total proteins in each transduced cell line compared with the cell line with the empty vector. (C) Validation of RPPA data for some proteins by immunoblot analysis. Protein extracts were subjected to immunoblot assays. To cover all of these proteins, 5 separate immunoblots were prepared, with actin used as the loading control for each. (D) The top 10 canonical pathways identified with Ingenuity Pathway Analysis and associated with the indicated BTKs.
Functional protein profiling of WT and mutant-BTK–overexpressing MEC-1 cells. Protein was extracted in biological triplicates from exponentially growing MEC-1 cells with either WT or mutant BTK and subjected to RPPA assays that included 426 antibodies. (A) Representative heat map of proteins with increased and decreased expression in each cell line (n = 3 per cell line). All BTK-overexpressing cell lines (BTKWT, BTKC481S, and BTKC481R) were compared with the BTKEV cell line. The fold changes of mean linear values were used to generate the heat map (fold change, ≥1.2 or ≤0.8). The color key indicates the fold change. (B) The number of increased (red) and decreased (blue) phosphorylated and total proteins in each transduced cell line compared with the cell line with the empty vector. (C) Validation of RPPA data for some proteins by immunoblot analysis. Protein extracts were subjected to immunoblot assays. To cover all of these proteins, 5 separate immunoblots were prepared, with actin used as the loading control for each. (D) The top 10 canonical pathways identified with Ingenuity Pathway Analysis and associated with the indicated BTKs.
Next, we compared BTKWT cells with mutant-BTK–expressing cells, as these 3 cell lines overexpress BTK. There were 22 upregulated and 23 downregulated transcripts in BTKC481S cells compared with BTKWT cells (Figure 2A-B). In contrast, the comparison of BTKWT and BTKC481R cells identified only 8 upregulated and 7 downregulated transcripts, indicating the similarity of these 2 cell lines at the transcriptomic level (Figure 2C-D). There were 2 DEGs (FYB and PKIB) that were upregulated and 3 (COL5A3, ABCC12, and NBEA) that were downregulated in both BTKC481S and BTKC481R cells compared with BTKWT cells. A comparison of the 2 resistant variants (BTKC481S vs BTKC481R) showed 46 upregulated and 20 downregulated DEGs (Figure 2E-F). The top canonical pathways that were identified in these comparisons are provided in supplemental Figure 3. These data demonstrate that all of the generated cell lines presented distinct molecular profiles at the transcript level; however the BTK overexpression rather than BTK mutations had a profound effect on the MEC-1 cell biology.
Functional protein profiling for WT and mutant-BTK–overexpressing MEC-1 cells
We first compared RPPA data (426 proteins) in all BTK-overexpressing cell lines (BTKWT, BTKC481S, and BTKC481R) to data from the BTKEV cell line. Fold changes of the linear expression values were used to generate the representative heat map in Figure 3A (cutoff fold changes, >1.2 or <0.8). The canonical pathways associated with each comparison are provided in supplemental Figure 4.
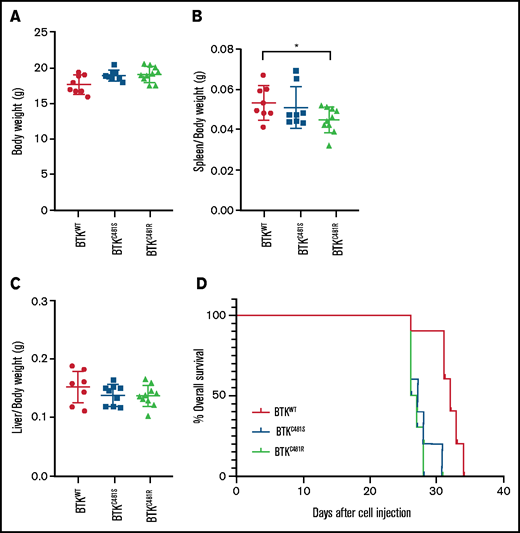
Comparison of in vivo parameters and tumor development in WT and mutant-BTK–expressing MEC-1 cells. MEC-1 cells harboring WT or mutant BTK were injected into 8-week-old Rag2−/−γc−/− female mice (n = 10 per group), and the animals were monitored for body weight and development and progression of leukemia. At the end point of the study, the spleens, livers, and femurs were collected and macroscopically evaluated. (A) Body weight of mice (n = 10 per group) in each cohort. (B) Spleen/body weight ratios of mice in each cohort (n = 8 BTKWT, n = 9 BTKC481S, n = 10 BTKC481R; *P < .05). (C) Liver/body weight ratios of mice in each cohort (n = 8 BTKWT, n = 9 BTKC481S, n = 10 BTKC481R). Groups were compared by fitting a linear mixed-effect model for analysis of variance. (D) Kaplan-Meier curves depict the overall survival of mice in each group (n = 10 per group; mutant vs WT, log-rank test; P = .0001).
Comparison of in vivo parameters and tumor development in WT and mutant-BTK–expressing MEC-1 cells. MEC-1 cells harboring WT or mutant BTK were injected into 8-week-old Rag2−/−γc−/− female mice (n = 10 per group), and the animals were monitored for body weight and development and progression of leukemia. At the end point of the study, the spleens, livers, and femurs were collected and macroscopically evaluated. (A) Body weight of mice (n = 10 per group) in each cohort. (B) Spleen/body weight ratios of mice in each cohort (n = 8 BTKWT, n = 9 BTKC481S, n = 10 BTKC481R; *P < .05). (C) Liver/body weight ratios of mice in each cohort (n = 8 BTKWT, n = 9 BTKC481S, n = 10 BTKC481R). Groups were compared by fitting a linear mixed-effect model for analysis of variance. (D) Kaplan-Meier curves depict the overall survival of mice in each group (n = 10 per group; mutant vs WT, log-rank test; P = .0001).
Seven proteins (COL6A1, FOXO3, HES1, PAR, PDCD1, STAT3, and RPS6_pS240/244) showed changes in all 3 cell lines (BTKWT, BTKC481S, and BTKC481R) relative to the BTKEV cell line (Figure 3B). Among these targets, we were able to validate PDCD1 (PD-1) and Gab2 (Figure 3C). In addition, our immunoblot results showed that p-Pyk2 (Y402), Lyn, and glycogen synthase levels were higher in all BTK-overexpressing cell lines than in parental MEC-1 cells and BTKEV cells.
Next, we compared the BTK-overexpressing cell lines. The top 10 canonical pathways associated with these comparisons were identified with Ingenuity Pathway Analysis and analyzed using the log2 ratios (Figure 3D). The comparison of the BTKWT and BTKC481S cells identified the top 3 canonical pathways as those for IL-15 production (IGF1R, INSR, PTK2, and Syk), BCR signaling (Gab2, PTK2, PTK2B, PTPRC, and Syk), and ERK/MAPK signaling (DUSP4, EIF4EBP1, PAK1, PTK2, and PTK2B). The top 10 canonical pathways in BTKWT relative to BTKC481R cells include chemokine signaling (MAPK14, PRKCA, PTK2, and PTK2B), BCR signaling (Gab2, MAPK14, PTK2, and PTK2B) and ERK/MAPK signaling (DUSP4, PRKCA, PTK2, and PTK2B). When we compared the 2 cell lines expressing the variants BTKC481S and BTKC481R, the top 3 canonical pathways were those for CD28 signaling in T-helper cells (CSK, PAK1, PIK3CA, CD45, and Syk), PI3K/AKT signaling (FOXO3, Gab2, GYS1, MCL1, and PIK3CA), and IL-3 signaling (Gab2, PAK1, PIK3CA, and PRKCA).
To integrate protein and messenger RNA (mRNA) data, we compared those 2 parameters and identified 7 targets (SMAD1, FOXO3, CCNE1, INSR, HES1, CD38, and PHGDH) that were significantly correlated in all of the cell lines compared (supplemental Figure 5). Taken together, these data demonstrate that all of the generated cell lines presented distinct molecular profiles at the transcript and protein levels.
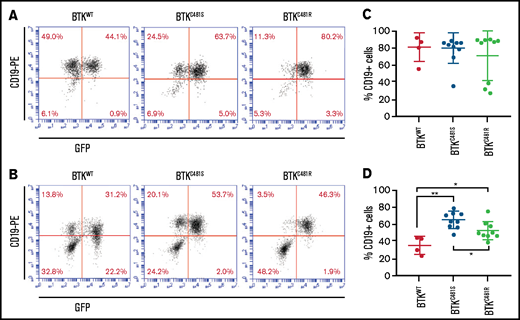
Localization of GFP+ and mutant-BTK–expressing MEC-1 cells in mice. Tumors were established from MEC-1 cells harboring WT or mutant BTK, as described in Figure 4. (A-B) Representative flow cytometry plots for the spleen and bone marrow samples collected from mice with BTKWT (n = 4; left), BTKC481S (n = 9; center), and BTKC481R (n = 9; right). At the time of euthanization, cells were collected from the spleen (A) and bone marrow (B), stained with a monoclonal antibody against human CD19 to identify the GFP+ MEC-1 cells, and analyzed by flow cytometry. (C-D) The percentage of CD19+ MEC-1 cells in spleen (C) and bone marrow (D). One-way analysis of variance, P = .05; Student t test, *P < .05 for BTKWT vs BTKC481R cells; **P < .01 for BTKWT vs BTKC481S cells.
Localization of GFP+ and mutant-BTK–expressing MEC-1 cells in mice. Tumors were established from MEC-1 cells harboring WT or mutant BTK, as described in Figure 4. (A-B) Representative flow cytometry plots for the spleen and bone marrow samples collected from mice with BTKWT (n = 4; left), BTKC481S (n = 9; center), and BTKC481R (n = 9; right). At the time of euthanization, cells were collected from the spleen (A) and bone marrow (B), stained with a monoclonal antibody against human CD19 to identify the GFP+ MEC-1 cells, and analyzed by flow cytometry. (C-D) The percentage of CD19+ MEC-1 cells in spleen (C) and bone marrow (D). One-way analysis of variance, P = .05; Student t test, *P < .05 for BTKWT vs BTKC481R cells; **P < .01 for BTKWT vs BTKC481S cells.
Establishment of and tumor development in xenograft mouse models harboring BTKWT, as well as BTKC481S and BTKC481R mutants
For the in vivo studies, we used transduced MEC-1 cell lines to establish a xenograft mouse model engrafted into Rag2−/−γc−/− mice. This model mimics aggressive CLL.25 After injection with one of the transduced MEC-1 cell lines, mice were monitored daily for development and progression of leukemia. We detected a loss of body weight in all groups ∽2 weeks after cell inoculation. After a month, all mice had developed CLL-like disease with systemic involvement (engraftment efficiency, 100%). Individual mice were euthanized when they became moribund and on the advice of our Veterinary Medicine Core service. At this time, the body weight, spleen, liver, peripheral blood and left femur of each mouse were collected. There were no statistically significant differences in body weight among the groups (Figure 4A). We observed splenomegaly, an increased number of localizations in the spleens and livers (macroscopic image shown later in Figure 7), and macroscopically evident areas of necrosis. BTKC481R group had the lowest spleen/body weight ratio, but there were no statistically significant differences in liver weights (P < .05; Figure 4B-C). The duration of overall survival was slightly shorter in mice that were injected with MEC-1 cells harboring mutant BTK (log-rank test; P = .0001; Figure 4D).
Localization of GFP+ WT and mutant-BTK–expressing cells in mice
Flow cytometric studies confirmed leukemic expansion of CD19+GFP+ cells in the spleen (Figure 5A), bone marrow (Figure 5B), and peripheral blood (data not shown). Because we had observed that GFP+ cells could lose GFP expression in vitro over time, we assessed the levels of CD19+ cells in the spleen and bone marrow samples to identify the leukemic B-cell populations. We found substantial accumulations of CD19+ cells in the spleens of all of the mice (Figure 5C); there were no significant differences among the 3 subtypes. However, in the bone marrow analysis, the percentage of mutant BTK cells was greater than that of the BTKWT cells; the greatest localization in all groups was of BTKC481S cells (Figure 5D). Infiltration of MEC-1 cells was observed in the spleen, liver, and bone (Figure 6) of all groups overexpressing BTK. Data from the morphologic diagnosis of organs and a summary of the mean scores for each evaluation are provided in supplemental Table 3.

Histopathological assessment of tissues. Histopathologic assessments of hematoxylin and eosin–stained slides were conducted by a veterinary pathologist. Representative images of tissues and lesions of lymphocytic infiltration were captured at the indicated magnifications for the main and inset images. The evaluation results, including the morphologic diagnoses of the organs and the summaries of the mean scores, are provided in supplemental Table 3. Liver and spleen: n = 8/no disease, n = 8/EV, n = 8/BTKWT, n = 9/BTKC481S, and n = 9/BTKC481R. Bone: n = 8/no disease, n = 6/EV, n = 7/BTKWT, n = 6/BTKC481S, and n = 6/BTKC481R.
Histopathological assessment of tissues. Histopathologic assessments of hematoxylin and eosin–stained slides were conducted by a veterinary pathologist. Representative images of tissues and lesions of lymphocytic infiltration were captured at the indicated magnifications for the main and inset images. The evaluation results, including the morphologic diagnoses of the organs and the summaries of the mean scores, are provided in supplemental Table 3. Liver and spleen: n = 8/no disease, n = 8/EV, n = 8/BTKWT, n = 9/BTKC481S, and n = 9/BTKC481R. Bone: n = 8/no disease, n = 6/EV, n = 7/BTKWT, n = 6/BTKC481S, and n = 6/BTKC481R.
Histopathology of lymphoid and nonlymphoid tissues in the xenograft system
Compared with the mice in the no-disease group, the remaining 3 groups showed similar spleen sizes at the end point of the study (Figure 7). Fluorescence microscopy showed that there were extensive accumulations of GFP+ MEC-1 cells in the spleens of the mice from the 3 BTK-overexpressing groups, and a higher number of nodules was also observed in those mice (supplemental Figure 6A-B). The cells from the spleens of all groups showed homogenous immunoreactivity for both human CD19 and human CD20, indicating the presence of transplanted MEC-1 cells, given that Rag2−/−γc−/− mice lack B cells. In addition, all evaluated spleens contained Ki67+ cells, suggesting high MEC-1 cell proliferation rates in all groups.
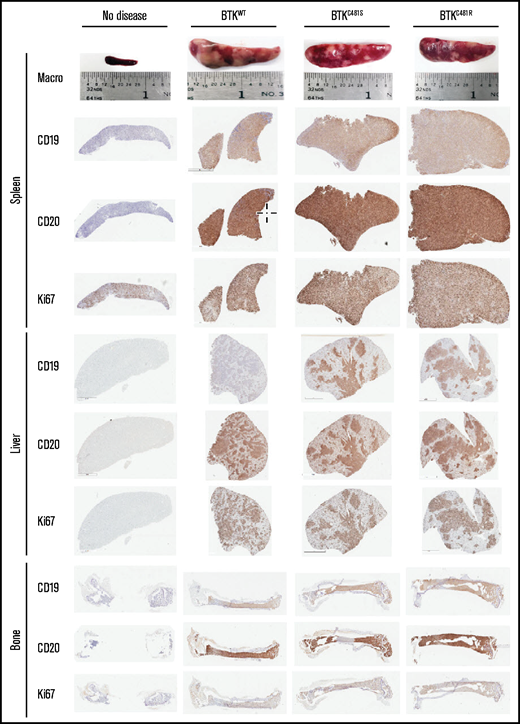
Immunohistochemical analyses of spleen, liver, and bone tissues after the establishment of disease from MEC-1 cells with WT or mutant BTK. Representative macroscopic images of spleens from each group are provided in the top row. Spleen, liver, and bone tissues were collected at the time of euthanasia. B-cell marker expression (CD19 and CD20) and proliferation marker expression (Ki-67) are shown for the spleen, liver, and bone marrow from mice with no disease and from BTKWT, BTKC481S, and BTKC481R mice.
Immunohistochemical analyses of spleen, liver, and bone tissues after the establishment of disease from MEC-1 cells with WT or mutant BTK. Representative macroscopic images of spleens from each group are provided in the top row. Spleen, liver, and bone tissues were collected at the time of euthanasia. B-cell marker expression (CD19 and CD20) and proliferation marker expression (Ki-67) are shown for the spleen, liver, and bone marrow from mice with no disease and from BTKWT, BTKC481S, and BTKC481R mice.
In contrast to the splenic tissues, the liver tissues in all groups showed localized expression of CD19+ and CD20+ MEC-1 cells (Figure 7). Likewise, these cells were equally proliferative, as indicated by their degree of Ki67 positivity. Similar observations were made in the bone marrow (Figure 7). To assess Ki67 positivity quantitatively, all of the liver slides were scanned with a digital scanner. The livers from all groups, except the no-disease group, showed ∽70% Ki67 positivity. There was no statistically significant difference among the groups overexpressing WT or mutant BTK (supplemental Figure 7). In addition, the splenic and liver tissues (n = 3 per group) were stained with p-S6 antibody, and the staining intensity was quantified. The intensity of p-S6 was slightly higher in the splenic tissues from the groups with mutant BTK than in the BTKWT group (supplemental Figure 8A), whereas it was similar in the liver tissues from all groups (supplemental Figure 8B).
Discussion
Point mutations in BTK at C481 prevent drug binding, which leads to ibrutinib-resistant clones. The absence of in vitro and in vivo models harboring these clones has left unanswered questions about the biology of ibrutinib resistance and patients’ responses to new agents that are effective against these resistant clones. Accordingly, in this study, we established in vitro and in vivo CLL models that harbor either overexpressed WT BTK or BTK point mutations (C481S or C481R) that mimic those observed in patients with ibrutinib-resistant CLL.
The impact of BTK mutation on leukemic cell proliferation was not previously known. In our study, several indicators suggested that the rate of proliferation did not differ in cells harboring either mutant or WT BTK. The growth rate and doubling time of these cells were similar in culture, although BTKC481R cells grew slightly more slowly. The cell cycle profiles were comparable. The proliferative index, determined by the level of the Ki67+ cell count, further demonstrated that, in the liver, the in vivo proliferation of all 4 BTK subtypes was similar. Collectively, these data suggest that overexpression of WT BTK or mutant BTKC481S/C481R does not result in the increased proliferation of cells.
Mouse models are powerful tools for investigating the biology of and therapeutic responses to novel agents. The most remarkable characteristic of CLL murine models is the penetrance of the CLL phenotype. Eμ-TCL1 was the first transgenic mouse model of CLL-like disease,27,28 and it has the highest penetrance (∽100%) of all murine CLL models29 and has been used to test numerous novel therapeutic strategies.30 In a recent study, researchers generated a double-transgenic mouse model (Eμ-TCL1xMyc) to study therapeutic strategies in concurrent CLL and B-cell lymphoma.31 However, the Eμ-TCL1 model typically requires 6 months for the appearance of circulating tumor cells.29 In contrast, xenotransplantation of MEC-1 cells provides a more immediate preclinical tool. Our mouse model developed disease in 1 month with 100% engraftment efficiency, which would allow for the efficacy of inhibitors to be determined in a 30-day period. Further, the use of engineered MEC-1 cell lines enabled us to have not only an in vivo model that mimics ibrutinib-resistant CLL but also isogenic cell lines for in vitro use.
CLL cells in humans reside in the bone marrow, lymph nodes, and peripheral blood,4 whereas in mice they are primarily in the spleen. The spleen size and density changed dramatically in the diseased compared with the disease-free animals; however, spleen size was similar among the BTKWT, BTKC481R, and BTKC481S mice. The spleen/body weight ratio in the BTKC481R mice was significantly lower than that in the BTKWT mice; this result may be attributable to the slightly lower proliferation rate of BTKC481R cells compared with BTKWT cells. In the spleens, the number of CD19+ cells per 100 000 cells was similar in all groups, whereas in the bone marrow, there were significantly more CD19+ cells in the mutant groups than in the BTKWT group, and this factor may be contributory to the early demise of mice with mutant-BTK–harboring MEC-1 cells. There are 2 possible reasons for increased CD19+ cells: either the cells harboring mutant BTK proliferated more in the bone marrow or these cells interacted better with the microenvironment and remained in the bone marrow for a longer time. Our in vitro data on the proliferation rates of the cell lines do not support the first possibility. Thus, further studies are needed to explain the interaction between the BTK mutant cells and the microenvironment in vivo.
A significant benefit of studying the resistant clones in vitro was the ability to explore the molecular signatures associated with overexpression of this enzyme as well as the mutational status of the kinase. First, our focus was on the targets that were altered by BTK overexpression, regardless of BTK mutation status. It was clear from the omics data that mere overexpression of the enzyme resulted in profound changes in DEGs. Some of these changes could be explored further with respect to development of resistance. For instance, Gab2 increased in all BTK-overexpressing cell lines (BTKWT, BTKC481S, and BTKC481R) compared with BTKEV, per both our RNAseq and RPPA data, and the protein levels were validated by our immunoblot results. Acquired ibrutinib resistance from exposure to escalating doses of the drug resulted in increased BTK and Gab2 expression in the Namalwa B-cell lymphoma cell line.32 Gab2 expression has also been reported to be significantly higher in the bone marrow of patients with chronic myeloid leukemia than in controls.33 In addition, Kim and colleagues34 recently identified Gab2 as a previously unrecognized target that altered DNA methylation in Korean patients with CLL.
Next, we compared mutant-BTK–expressing cells to BTKWT cells. This comparison revealed 5 common DEGs (NBEA, COL5A3, ABCC12, FYB, and PKIB) that were altered in mutant compared with BTKWT cells. We also identified targets that were previously reported to be associated with CLL or other hematologic malignancies. For instance, EphA3, GNAQ, and GATA3 mRNA levels were higher in BTKC481S than in BTKWT cells. Previously, an aberrant EphA3 copy number was found to be associated with multiple types of hematologic malignancies,35,36 and mRNA levels correlated positively with the copy numbers.36 Recently, driver mutations in GNAQ and GATA3 have been identified in a patient with CLL.37-39 We observed that GATA3 transcript and protein levels were higher in BTKC481S than in BTKWT cells; however, the levels in patients with CLL remain unknown, and the clinical outcomes associated with the expression levels of these targets should be studied.
In the integrative analyses of RNAseq and RPPA results, only 7 genes had adjusted P < .05 (SMAD1, FOXO3, CCNE1, INSR, HES1, CD38, and PHGDH). It is important to underscore that there are only 426 proteins in this RPPA panel. SMAD1 expression was lower in all BTK-overexpressing cell lines at both the transcript and protein levels. It has been shown that the TGF-β-SMAD pathway is inactivated in CLL40 and has a role in follicular lymphoma.41 In addition, SMAD1 expression is aberrantly downregulated in >85% of patients with diffuse large B-cell lymphoma and provides a proliferative advantage to B cells in vitro and in vivo.42
Our transcriptomic and proteomic data delineated molecular differences between WT and BTK-mutant MEC-1 cells. Importantly, between the 2 variants (C481S and C481R), we identified differential expression of both proteins and transcripts. Collectively, our data confirm the distinct molecular signatures of these subclones that may lead to context-specific resistance and further the development of personalized medicine and potential combination strategies.
A limitation of our study is the lack of validation of our findings in patients harboring BTK subclones. The primary reason for this is that there are insufficient numbers of purified CLL cells from patients with these mutations. Therefore, further evaluation with clinical data are needed for a comprehensive characterization. Nonetheless, our current work is of value because it established in vitro and in vivo ibrutinib-resistant prototypes harboring the most prominent BTK C481 mutations or overexpression of WT BTK. Because the engraftment, expansion, and dissemination of these BTK cells in lymphoid and nonlymphoid tissues mimics human CLL, our models may be valuable preclinical tools for understanding the pathophysiology of the disease and testing a new generation of reversible BTK inhibitors that do not require C481 binding. These new-generation BTK inhibitors include Loxo-305,43,44 vecabrutinib,45-47 and ARQ 531,48 which bind to an allosteric site of the kinase and inhibit BTK and its downstream signaling. These agents are in early clinical development for patients with relapsed/refractory CLL and mantle cell lymphoma and are primarily intended for those in whom therapy with irreversible BTK inhibitors has failed. Currently, we are testing a reversible BTK inhibitor in this model system. In addition, our in vitro and in vivo models may provide a tool for evaluating combination strategies that could be tested and validated in cell lines and animals before clinical use. Finally, our modified MEC-1 cell line and xenograft mouse models could be a context-specific model system applicable to other C481 or PLCγ2.
Acknowledgments
The authors thank Laura L. Russell of MD Anderson Cancer Center’s Research Medical Library for editing the manuscript.
This work was supported in part by The University of Texas MD Anderson Cancer Center Moon Shot Program; by National Institutes of Health, National Cancer Institute grant P30CA016672 (the Flow Cytometry and Cellular Imaging Core Facility, Functional Proteomics RPPA Core Facility, The Sequencing and Non-Coding Program Laboratory, Advanced Technology Genomics Core Cytogenetics, Cell Authentication Core Facility Research Animal Support Facility-Houston, Research Histology Core Laboratory, and Department of Veterinary Medicine and Surgery); and by the Scientific and Technological Research Council of Turkey (TÜBİTAK) Science Fellowships and Grant Programs (BIDEB) International Postdoctoral Research Fellowship Program 2219 (G.K.).
Authorship
Contribution: B.A. designed and performed the experiments, analyzed the results, and wrote the first draft of the manuscript; G.K. assisted in all in vivo experiments and in planning the work; L.S.C. and L.R.I. assisted at the end point of the in vivo experiments; M.M. and M.P. worked on the construction of the plasmids for all of the BTK clones and the establishment of cell lines following transduction; M.G. evaluated all of the pathology results and contributed to the writing of the manuscript; N.W.F. captured images of the cells with a fluorescence microscope; X.Z. performed all of the bioinformatics analyses of the RPPA and RNAseq data; J.W. supervised X.Z.; C.P.V. supervised M.M. and M.P.; J.R.M. supervised M.M. and M.P., participated in intellectual discussions regarding model systems, and wrote and reviewed the manuscript; M.T.S.B. directed the in vivo experiments for the development of the mouse models and wrote and reviewed the manuscript; and V.G. conceptualized and supervised the research, obtained funding, analyzed the data, and wrote and reviewed the manuscript.
Conflict-of-interest disclosure: V.G. has sponsored research agreements from Sunesis and Loxo Oncology. The remaining authors declare no competing financial interests.
The current affiliation for G.K. is Department of Biochemistry, Ankara University Faculty of Veterinary Medicine, Ankara, Turkey.
Correspondence: Varsha Gandhi, Department of Experimental Therapeutics, The University of Texas MD Anderson Cancer Center, Unit 1950, 1901 East Rd, Houston, TX 77054; e-mail: vgandhi@mdanderson.org.

