

Key Points
Thrombin and the viral RNA analogue poly(I:C) cooperatively amplify endothelial prothrombotic and proinflammatory function.
Inhibition of PAR2 reduces the expression of tissue factor and leukocyte recruitment by endothelial cells.

Abstract
Activation of blood coagulation and endothelial inflammation are hallmarks of respiratory infections with RNA viruses that contribute significantly to the morbidity and mortality of patients with severe disease. We investigated how signaling by coagulation proteases affects the quality and extent of the response to the TLR3-ligand poly(I:C) in human endothelial cells. Genome-wide RNA profiling documented additive and synergistic effects of thrombin and poly(I:C) on the expression level of many genes. The most significantly active genes exhibiting synergistic induction by costimulation with thrombin and poly(I:C) included the key mediators of 2 critical biological mechanisms known to promote endothelial thromboinflammatory functions: the initiation of blood coagulation by tissue factor and the control of leukocyte trafficking by the endothelial-leukocyte adhesion receptors E-selectin (gene symbol, SELE) and VCAM1, and the cytokines and chemokines CXCL8, IL-6, CXCL2, and CCL20. Mechanistic studies have indicated that synergistic costimulation with thrombin and poly(I:C) requires proteolytic activation of protease-activated receptor 1 (PAR1) by thrombin and transactivation of PAR2 by the PAR1-tethered ligand. Accordingly, a small-molecule PAR2 inhibitor suppressed poly(I:C)/thrombin–induced leukocyte-endothelial adhesion, cytokine production, and endothelial tissue factor expression. In summary, this study describes a positive feedback mechanism by which thrombin sustains and amplifies the prothrombotic and proinflammatory function of endothelial cells exposed to the viral RNA analogue, poly(I:C) via activation of PAR1/2.
Introduction
Activation of blood coagulation is invariably linked to the innate immune response to infection by viral and bacterial pathogens, secondary to augmented expression of the initiator of the extrinsic pathway of blood coagulation, tissue factor (TF; gene symbol, F3) on innate immune cells and vascular endothelial cells (ECs).1-3 Aberrant coagulation activation and thrombosis have been recognized as a contributing factor in the pathology of respiratory tract infections with influenza A viruses, Middle East respiratory syndrome, and severe acute respiratory syndrome coronavirus (SARS-CoV1 and -2).4-6 The thrombotic coagulopathy affecting the pulmonary circulation and secondary organs such as the liver and kidneys of patients with COVID-19,7-14 together with early clinical observations indicating a potential benefit of anticoagulant interventions,15-17 suggest that dysregulated coagulation contributes significantly to the morbidity and mortality of patients with severe disease.
The extent of coagulopathy triggered by single-stranded RNA viruses has led to suggestions that the acute thrombotic pathology associated with respiratory tract infection may in part be caused by excessive EC injury and inflammatory activation.18-21 This state of endothelial activation comprises wide-ranging adaptations that support a localized immune response by facilitating leukocyte trafficking across the blood-tissue barrier, controlling blood supply to sites of infections, regulating blood pressure, and promoting the localized activation of platelets and the blood coagulation mechanism. Dysregulation of these responses caused by excessive, sustained elaboration of proinflammatory mediators and cytokines, as it occurs in systemic inflammatory response syndrome and severe sepsis, has been linked to life-threatening failure to sustain adequate blood pressure, microvascular thrombosis, and, in the most severe cases, to disseminated intravascular coagulation and multiorgan failure.
The TF/FVIIa complex–initiated activation of the coagulation proteases factor VII and X and the ensuing downstream generation of thrombin not only trigger the procoagulant state associated with infection, but in addition may modulate cellular functions via G-protein–coupled protease-activated receptors (PARs) 1, 2, and 4 (reviewed in Posma et al22 and Samad and Ruf 23). Experimental evidence indicates that thrombin signaling via PARs alters the function of human ECs in a manner similar to inflammatory cytokines, including increased leukocyte trafficking, permeability, vasomotor tone, angiogenesis, and TF expression.24-27
The role of direct endothelial infection by viral pathogens remains to be fully explored. For example, ECs express the primary receptor for SARS-Cov1/2 and angiotensin-converting enzyme 2, and elevated endothelial angiotensin-converting enzyme 2 is associated with the cardiovascular risk factors predictive of increased morbidity.28,29 SARS-CoV-2 RNA has been detected in the peripheral blood of some patients with severe disease30 and the virus infects ECs in vitro31 and in vivo.18,32 A significant role for ECs as the source of procoagulant activity and cytokine production induced by viral infection is further suggested by the observation that the viral RNA analogue polyinosinic:polycytidylic acid (poly[I:C]) induces both cytokine production and TF-procoagulant activity via Toll-like receptor 3 (TLR3) in human umbilical vein ECs (HUVECs). In contrast, poly(I:C) induced the release of cytokines, but not TF expression in human peripheral blood-derived monocytes.33
In the current work, we investigated how signaling by TF and activated coagulation proteases affects the EC response to the viral RNA analogue and TLR3-ligand poly(I:C).
Materials and methods
Cell culture
EA.hy926 cells (CRL-2922; ATCC) were cultured in Dulbecco’s modified Eagle’s medium with 20 mM HEPES, 4 mM glutamine, 1 mM sodium pyruvate, 0.75 g/L sodium bicarbonate, 100 U/mL penicillin, 100 μg/mL streptomycin, and 10% fetal bovine serum. Pooled HUVECs (cat. no. C2517A; Lonza, Walkersville, MD) were cultured in endothelial basal medium (cat. no. CC-3162; Lonza), containing 1 μg/mL hydrocortisone, 10 ng/mL epidermal growth factor, 10 ng/mL basic fibroblast growth factor, and 5% (v/v) fetal calf serum. THP-1 cells (TIB-202; ATCC) were cultured in RPMI 1640 (Life Technologies, Grand Island, NY) supplemented with 10% fetal bovine serum, 10 mM HEPES, 0.1 mM minimum essential medium nonessential amino acids, 1 mM sodium pyruvate, and 100 nM penicillin/streptomycin (Life Technologies), in 5% CO2 at 37°C.
TF activity
Total and cell surface TF activity were measured by 1-stage clotting assays and 2-stage fXa generation assays, respectively, as described,33 with the anti-TF antibody HTF-134 (10 µg/mL). Clotting times were converted into procoagulant activity from a standard curve generated with human TF (Innovin; Dade Behring, Germany). The PAR1 and PAR2 cleavage-blocking antibodies ATAP2 (10 μg/mL), WEDE15 (10 μg/mL), SAM-11 (10 μg/mL), vorapaxar (1 μg/mL), and GB83 (25 µM) were added 1 hour before poly(I:C) and/or thrombin.
Quantitative reverse transcription‒real-time PCR
Total RNA was extracted with TRIzol (Life Technologies, Carlsbad, CA) and transcribed into cDNA using the QuantiTect Reverse transcription kit (Qiagen), and transcript levels were quantified by TaqMan PCR assays with commercially available primer sets (Applied Biosystems), by the ΔΔCt method, with glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as a normalizing control.35
RNA sequencing
cDNA libraries were prepared from poly(A)-selected RNA (NEBNext Poly[A] messenger RNA [mRNA] magnetic isolation kit) with the NEBNext Ultra RNA Library Prep Kit (cat. no. E7530; NEB) and sequenced with the Illumina NextSeq 500 system (75 cycles). Sequence data were demultiplexed by using bcl2fastq v2.20.0.422 and aligned and quantitated with ARMOR pipeline v1.2.236 with Ensemble Human genome GRCh38 release 93 as the reference. Expression quantifications were imported into R v3.5.3 and summarized at the gene level by using tximport v1.10.0. All samples were normalized, and gene expression was compared between conditions with Wald tests in DESeq2 v1.22.1. P-values were corrected for multiple tests by the Benjamini-Hochberg method. Log-fold changes were shrunk by using the normal method in DESeq2. Genes with an adjusted P < .05 (Padj.) were considered to be significantly differentially expressed. Differentially expressed genes were annotated using the Gene Ontology database (https://biit.cs.ut.ee/gprofiler) to examine the biological functions and pathways of the genes.
Western blot analysis
ECs were lysed in RIPA buffer (Invitrogen, Rockford, IL), subjected to electrophoresis on 10% sodium dodecyl sulfate-polyacrylamide gels (Mini-PROTEAN TGX; Bio-Rad) under reducing conditions, and transferred to 0.2-µm PVDF membranes on a Trans-blot TURBO transfer system (Bio-Rad). Membranes were probed with mouse anti-human TF (clone, VIC7; Sekisui Diagnostics, Darmstadt, Germany) and mouse anti-human β-tubulin (clone, BT7R; Invitrogen) primary antibodies, followed by horseradish peroxidase (HRP)-conjugated secondary antibodies, and imaged on an Amersham Imager 680 (General Electric). Band density was quantified with ImageJ software (National Institutes of Health, http://rsb.info.nih.gov/ij/).
Immunofluorescence microscopy
Cells were cultured on coverslips, fixed for 1 hour at 4°C in phosphate-buffered saline (PBS)-4% paraformaldehyde, permeabilized with 0.1% Triton X-100 in PBS for 5 minutes when indicated, blocked with 2% bovine serum albumin-PBS for 1 hour, incubated overnight at 4°C with mouse anti-human TF mAb (clone VIC7; 1:1000) or isotype-control followed by FITC-conjugated anti-mouse IgG for 60 minutes, mounted in 4′,6-diamidino-2-phenylindole medium (ProLong Gold antifade reagent; Invitrogen), and imaged on a Nikon Eclipse Ti2 inverted microscope at ×60 original magnification. Images of nonpermeabilized cells were acquired on a FV1000-MPE Laser Scanning Confocal and Multiphoton Microscope (Olympus) at ×100 original magnification .
Generation of Thbd-deficient EA.hy926 cells
Guide RNA sequences targeting the 5'- and 3'-UTRs of the thrombomodulin (Thbd) gene (5'-UTR target: 5'-GCAGGCGCCGGGGAAAGCGC-3'; 3'-untranslated region (UTR) target: 5'-TGAATTTGGATATCTCGCAG-3') were cloned into the Cas9 expression plasmid pX459.V2 (cat. no. 62988; Addgene), and the recombinant plasmids were cotransfected into EA.hy926 cells by nucleofection (Cell Line Optimization 4D-Nucleofector X Kit; Lonza). After expansion, Thbd-deficient cells were enriched by fluorescence-activated cell sorting with phycoerythrin (PE)-conjugated anti-Thbd antibodies (CD141/BDCA-3-PE, cat. no. 130-090-514; Miltenyi Biotec), as a pooled population or by sorting single Thbd− cells into 96 wells, followed by expansion and validation of Thbd negativity by flow cytometry and PCR amplification/sequencing of the modified Thbd locus (“single-cell clone”).
Flow cytometry
Single-cell suspensions prepared with cell dissociation buffer (Gibco) were incubated with PE-conjugated mouse anti-human E-selectin, APC-conjugated mouse anti-human VCAM1, or nonimmune isotype controls (cat. no. 322605; CD62E-PE; cat. no. 305809; CD106-APC, BioLegend; mouse IgG2a and IgG1; cat. no MA5-14453 and 6-4724-82; eBioscience) and analyzed on a BD Accuri C6 flow cytometer.
EC-leukocyte adhesion assay
HUVECs cultured in 12-well dishes were treated for 6 hours with poly(I:C) and/or thrombin and washed with PBS, and THP-1 cells labeled with rhodamine B isothiocyanate-dextran (Signa Aldrich) were added. After 1 hour, unbound cells were removed by 3 PBS washes, and bound cells were counted under a fluorescence microscope (Olympus BX-51). Vorapaxar (1 μg/mL) or GB83 (25 µM) was added 1 hour before the THP-1 cells, and VCAM1 and E-selectin–blocking antibodies (10 μg/mL) were added 30 minutes before the THP-1 cells.
Statistical analysis
Statistical analysis was performed with Prism 8 software (GraphPad) by 1-way analysis of variance (ANOVA) for multiple groups and the Bonferroni multiple-comparisons test or Student t test for 2 groups.
Results
Genome-wide transcriptome effects of costimulation with thrombin and poly(I:C)
Triplicate cultures of HUVECs were treated for 3 hours with poly(I:C) and/or thrombin and subjected to transcriptional profiling. Similar to that described earlier,33,37-39 poly(I:C)-induced transcriptome changes (66 genes significantly upregulated and 14 downregulated (up/down) >1.5-fold vs unstimulated cells; Padj < .05) included increased transcript levels for receptors and chemokines controlling leukocyte migration and adhesion, interferon-responsive transcripts, and genes associated with inflammatory endothelial activation (Figure 1; supplemental File 1). In line with independent studies,40,41 thrombin likewise induced a proinflammatory gene signature (143 up and 69 down >1.5-fold vs unstimulated cells; Padj < .05) that partially overlapped with the response to poly(I:C) (Figure 1; Venn section F). Genes within this overlapping subset regulate critical aspects of leukocyte trafficking (such as VCAM1, E-selectin [gene symbol: SELE], ICAM1, and CXCL1 and 8) and endothelial function in conditions of inflammatory stress (such as JUNB, NFKB2, RELB, CD69, and SOD2; Figure 1; supplemental File 1). Approximately half of the gene responses in costimulated cells were dominated by either thrombin or poly(I:C) (Figure 1; Venn sections E and G). For example, the expression of core interferon-responsive genes (such as APOL2; RIPK2; OASL; IFI44; IFIT family members 1, 2, and 3; and IRF1) was strongly induced by poly(I:C), but not by thrombin, and was sustained in costimulated cells at approximately the same level as in cells treated with poly(I:C) alone. Likewise, a large number of genes regulated by costimulation were selectively sensitive to thrombin, with little or no contribution of poly(I:C).
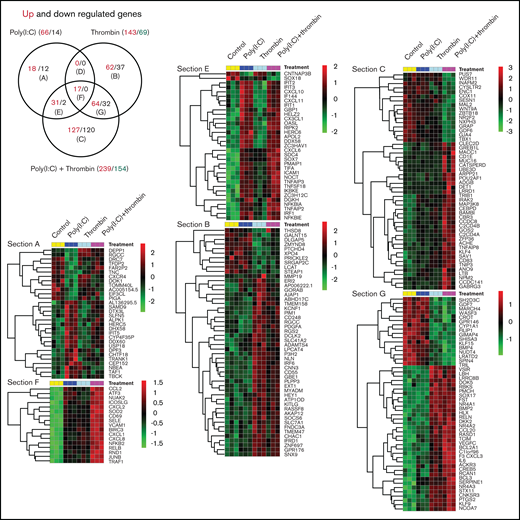
Transcriptional profiling of HUVEC responses to poly(I:C) and thrombin. (A) The Venn diagram shows the number of treatment-regulated genes in HUVECs in response to a 3-hour exposure to thrombin (5 nM), poly(I:C) (12.5 μg/mL), or both. Venn sections are labeled (A) to (G), with the number of upregulated (red font) and downregulated (black font) differentially expressed genes (>1.5-fold change, Padj. ≤ .05, measured with DESeq2 v1.22.1) indicated in each section. (B) Heat maps show the log2-fold differences in the abundance of all transcripts in sections E and F, and the most highly regulated transcripts in Venn sections G and C.
Transcriptional profiling of HUVEC responses to poly(I:C) and thrombin. (A) The Venn diagram shows the number of treatment-regulated genes in HUVECs in response to a 3-hour exposure to thrombin (5 nM), poly(I:C) (12.5 μg/mL), or both. Venn sections are labeled (A) to (G), with the number of upregulated (red font) and downregulated (black font) differentially expressed genes (>1.5-fold change, Padj. ≤ .05, measured with DESeq2 v1.22.1) indicated in each section. (B) Heat maps show the log2-fold differences in the abundance of all transcripts in sections E and F, and the most highly regulated transcripts in Venn sections G and C.
Notably, a large number of genes were significantly upregulated and downregulated only in costimulated cells (Figure 1; Venn section C). This pool included many genes with validated functions in ECs, such as ACHE, CEBPD, GDF6, C2CD4B, ADORA2B, and NEDD4,42–47 or with candidate functions established in non-EC types, such as CARD8, GBP2, REL, UGT8, and POU2AF1 (for a full list, see supplemental File 1). The results of an analysis of expression data for genes in Venn section F with Ingenuity Pathway Analysis tools and gene annotations of biological functions and pathways curated in the NCBI Gene Ontology database was consistent with a general transcriptional activation and a predominant effect on inflammatory responses (supplemental File 2; pathway analysis). The bias toward inflammatory endothelial activation was more pronounced in upregulated genes, whereas database-curated candidate endothelial functions associated with downregulated genes included angiogenesis, vasculogenesis, and vascular integrity.
Importantly, genes affected most by costimulation (fold up/down change in costimulated cells vs poly[I:C] alone, <2.5; Table 1) included TF (F3), cyclooxygenase 2 (PTGS2, alias COX2), E-selectin, VCAM1, interleukin-8 (CXCL-8), interleukin-6, chemokine (C-X-C motif) ligands-2/3 (CXCL2/3), C-C motif chemokine ligand-20 (CCL20), and the transcription factors NRA4A1 and 2 (alias NURR77/TR3 and NURR1), which are known to regulate critical endothelial functions in thrombus resolution,48 interactions with leukocytes,49 proliferation,50 microvascular barrier maintenance,51 and the inflammatory response to TNFα.52
Transcripts upregulated or downregulated ≥2.5-fold by costimulation with poly(I:C) and thrombin, relative to poly(I:C) alone
| Gene | Poly(I:C) + thrombin vs control | Poly(I:C) vs control | Thrombin vs control | Poly(I:C) + thrombin vs poly(I:C) | Poly(I:C) + thrombin vs thrombin |
|---|---|---|---|---|---|
| SELE | 13.12 | 3.85 | 4.14 | 3.41 | 3.17 |
| VCAM1 | 9.72 | 2.79 | 3.09 | 3.48 | 3.15 |
| CXCL8 | 6.66 | 1.88 | 3.4 | 3.54 | 1.96 |
| F3 | 6.47 | −1.33 | 2.36 | 8.59 | 2.74 |
| CXCL2 | 5.46 | 1.86 | 1.91 | 2.94 | 2.86 |
| NR4A1 | 4.32 | −1.52 | 4.42 | 6.53 | 0.98 |
| CXCL3 | 4.22 | 1.22 | 2.24 | 3.46 | 1.88 |
| IL6 | 3.14 | 1.12 | 1.8 | 2.8 | 1.74 |
| MUC16 | 3.03 | −1.2 | −1.28 | 3.64 | 3.88 |
| C11orf96 | 3.01 | −1.18 | 1.62 | 3.54 | 1.86 |
| CCL20 | 3.01 | 1.01 | 2.02 | 3 | 1.49 |
| PTGS2 | 2.85 | −1.02 | 2.43 | 2.9 | 1.17 |
| HLX | 2.83 | −1.06 | 3.23 | 3 | 0.88 |
| NR4A2 | 2.78 | −1.45 | 2.17 | 4.02 | 1.28 |
| FST | 2.68 | −1.16 | 2.92 | 3.1 | 0.92 |
| PMCH | 2.64 | −1.54 | 3 | 4.06 | 0.88 |
| RCAN1 | 2.57 | −1.11 | 2.08 | 2.85 | 1.24 |
| ADGB | 2.46 | −1.22 | −1.08 | 3.01 | 2.66 |
| BCL2A1 | 2.42 | −1.39 | 1.59 | 3.37 | 1.52 |
| UBE3D | 2.38 | −1.12 | −1.25 | 2.68 | 2.98 |
| DKK2 | 2.31 | −1.56 | 2.06 | 3.61 | 1.12 |
| CD1E | 2.1 | −1.22 | −1.22 | 2.57 | 2.56 |
| LBH | 1.8 | −1.43 | 2.59 | 2.56 | 0.69 |
| IL11 | 1.74 | −1.47 | 2.28 | 2.56 | 0.76 |
| CYSLTR2 | −1.96 | 1.31 | −1.39 | −2.58 | 1.41 |
| TRIL | −2.94 | −1.14 | −1.96 | −2.58 | 1.50 |
| Gene | Poly(I:C) + thrombin vs control | Poly(I:C) vs control | Thrombin vs control | Poly(I:C) + thrombin vs poly(I:C) | Poly(I:C) + thrombin vs thrombin |
|---|---|---|---|---|---|
| SELE | 13.12 | 3.85 | 4.14 | 3.41 | 3.17 |
| VCAM1 | 9.72 | 2.79 | 3.09 | 3.48 | 3.15 |
| CXCL8 | 6.66 | 1.88 | 3.4 | 3.54 | 1.96 |
| F3 | 6.47 | −1.33 | 2.36 | 8.59 | 2.74 |
| CXCL2 | 5.46 | 1.86 | 1.91 | 2.94 | 2.86 |
| NR4A1 | 4.32 | −1.52 | 4.42 | 6.53 | 0.98 |
| CXCL3 | 4.22 | 1.22 | 2.24 | 3.46 | 1.88 |
| IL6 | 3.14 | 1.12 | 1.8 | 2.8 | 1.74 |
| MUC16 | 3.03 | −1.2 | −1.28 | 3.64 | 3.88 |
| C11orf96 | 3.01 | −1.18 | 1.62 | 3.54 | 1.86 |
| CCL20 | 3.01 | 1.01 | 2.02 | 3 | 1.49 |
| PTGS2 | 2.85 | −1.02 | 2.43 | 2.9 | 1.17 |
| HLX | 2.83 | −1.06 | 3.23 | 3 | 0.88 |
| NR4A2 | 2.78 | −1.45 | 2.17 | 4.02 | 1.28 |
| FST | 2.68 | −1.16 | 2.92 | 3.1 | 0.92 |
| PMCH | 2.64 | −1.54 | 3 | 4.06 | 0.88 |
| RCAN1 | 2.57 | −1.11 | 2.08 | 2.85 | 1.24 |
| ADGB | 2.46 | −1.22 | −1.08 | 3.01 | 2.66 |
| BCL2A1 | 2.42 | −1.39 | 1.59 | 3.37 | 1.52 |
| UBE3D | 2.38 | −1.12 | −1.25 | 2.68 | 2.98 |
| DKK2 | 2.31 | −1.56 | 2.06 | 3.61 | 1.12 |
| CD1E | 2.1 | −1.22 | −1.22 | 2.57 | 2.56 |
| LBH | 1.8 | −1.43 | 2.59 | 2.56 | 0.69 |
| IL11 | 1.74 | −1.47 | 2.28 | 2.56 | 0.76 |
| CYSLTR2 | −1.96 | 1.31 | −1.39 | −2.58 | 1.41 |
| TRIL | −2.94 | −1.14 | −1.96 | −2.58 | 1.50 |
All genes were differentially expressed with an Padj. ≤ .05, measured with DESeq2 v1.22.1.
Overall, these analyses documented that costimulation with thrombin quantitatively and qualitatively altered the mRNA profile of ECs exposed to poly(I:C) and that biological mechanisms regulated most significantly at the mRNA level by costimulation included the initiation of blood coagulation and the control of leukocyte trafficking.
PAR1 or PAR2 activation amplifies poly(I:C)-induced TF and CXCL8 mRNA
EA.hy926 cells and HUVECs were treated with a combination of poly(I:C) and agonists for PAR1 (TFLLRN-NH2), PAR1/2 (SFLLRN-NH2), PAR2 (SLIGKV-NH2), PAR3 (TFRGAP-NH2), or PAR4 (GYPGV-NH2). In both cell types, poly(I:C) increased TF and CXCL8 mRNA abundance. Agonist peptides for PAR1 and PAR2, but not for PAR3 or PAR4, elicited a 4- to 10-fold increase in TF and CXCL8 mRNA in the absence of poly(I:C) and significantly augmented the poly(I:C) response (Figure 2).
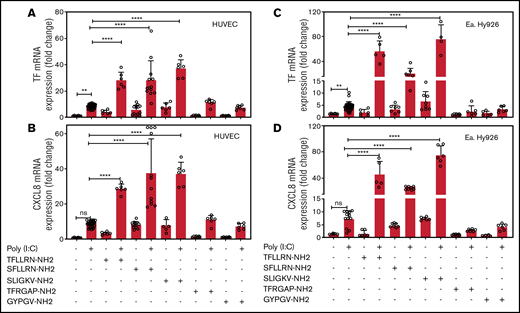
PAR1 and PAR2 activation augments poly(I:C)-induced expression of TF and CXCL8 mRNA. HUVECs and EA.hy926 cells were stimulated with poly(I:C) (12.5 μg/mL) and PAR-specific agonist peptides (150 μM; PAR-1, SFLLRN-NH2 and TFLLRN-NH2; PAR-2, SLIGKV-NH2; PAR-3, TFRGAP-NH2; and PAR-4, GYPGV-NH2) for 3 hours and TF (A,C) and CXCL8 (B,D) mRNA abundance relative to GAPDH were measured by RT-PCR (n = 6-18). Data represent the mean ± standard deviation of the target gene/GAPDH ratio relative to the control. ****P < .0001 by ANOVA followed by a multiple-comparison test. ns, not significant.
PAR1 and PAR2 activation augments poly(I:C)-induced expression of TF and CXCL8 mRNA. HUVECs and EA.hy926 cells were stimulated with poly(I:C) (12.5 μg/mL) and PAR-specific agonist peptides (150 μM; PAR-1, SFLLRN-NH2 and TFLLRN-NH2; PAR-2, SLIGKV-NH2; PAR-3, TFRGAP-NH2; and PAR-4, GYPGV-NH2) for 3 hours and TF (A,C) and CXCL8 (B,D) mRNA abundance relative to GAPDH were measured by RT-PCR (n = 6-18). Data represent the mean ± standard deviation of the target gene/GAPDH ratio relative to the control. ****P < .0001 by ANOVA followed by a multiple-comparison test. ns, not significant.
Thrombin amplifies poly(I:C)-induced TF and CXCL8 mRNA expression
HUVEC and EA.hy926 TF cell-surface activity was significantly increased after poly(I:C) stimulation (Figure 3A). Factors VIIa and fXa, the binary TF-fVIIa complex, and the ternary TF-fVIIa-Xa signaling complex had no effect on TF or CXCL8 mRNA in poly(I:C)-treated or unchallenged EA.hy926 cells (Figure 3B). In contrast, thrombin markedly amplified the poly(I:C)-induced increase of TF cell-surface activity over a dose range from 0.5 to 10 nM (Figure 3C).
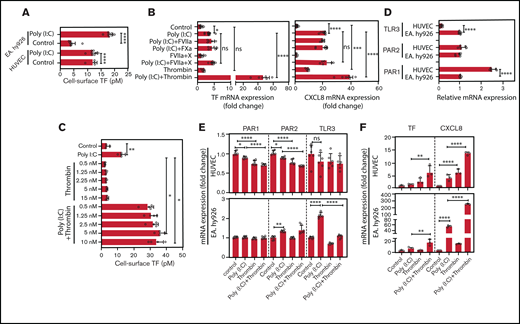
Role of thrombin and coagulation factors VII and X in the augmentation of poly(I:C)-induced responses EC. (A) HUVECs and EA.hy926 cells were stimulated with poly(I:C) (12.5 μg/mL) for 6 hours and the functionally active cell-surface TF was determined by FXa generation assays (n = 4-8). (B) EA.hy926 cells were stimulated with poly(I:C) (12.5 μg/mL) and candidate physiological PAR2 agonists, such as fXa (100 nM), fVIIa (FVIIa-100 nM), fVIIa plus fX (fVIIa, 500 pM; fX, 150 nM; enabling ternary TF-VIIa-Xa complex formation), or thrombin (5 nM) for 3 hours. Relative TF and CXCL8 mRNA abundance was determined by RT-PCR (n = 5-12 per condition). (C) EA.hy926 cells were pretreated with poly(I:C) (12.5 μg/mL) and thrombin (0.5-10 nM) for 6 hours, and the functionally active cell-surface TF was determined by an fXa-generation assay (n = 4). (D) Baseline abundance of TLR3, PAR1, and PAR2 mRNA relative to GAPDH mRNA in EA.hy926 cells and HUVECs. Target gene/GAPDH ratios in EA.hy926 cells were arbitrarily set to 1. (E) Changes in the abundance of PAR1, PAR2, and TLR3 mRNA relative to GAPDH mRNA in response to thrombin and/or poly(I:C) (n = 6) in EA.hy926 and HUVECs stimulated for 3 hours with poly(I:C) (12.5 μg/mL) and/or thrombin (5 nM). Baseline levels were arbitrarily set to 1. (F) TF and CXCL8 mRNA abundance in HUVECs and EA.hy926 cells treated for 3 hours with poly(I:C) (12.5 μg/mL) and/or thrombin (5 nM; n = 6/condition). All data represent the mean ± standard deviation of the indicated number of replicates and were generated in at least 2 biological replicates. The statistical significance of the differences between 2 groups was analyzed by Student t test. A comparison of more than 2 groups was conducted by ANOVA followed by a multiple-comparison test. *P < .05; **P < .01; ***P < .001; ****P < .0001. ns, nonsignificant.
Role of thrombin and coagulation factors VII and X in the augmentation of poly(I:C)-induced responses EC. (A) HUVECs and EA.hy926 cells were stimulated with poly(I:C) (12.5 μg/mL) for 6 hours and the functionally active cell-surface TF was determined by FXa generation assays (n = 4-8). (B) EA.hy926 cells were stimulated with poly(I:C) (12.5 μg/mL) and candidate physiological PAR2 agonists, such as fXa (100 nM), fVIIa (FVIIa-100 nM), fVIIa plus fX (fVIIa, 500 pM; fX, 150 nM; enabling ternary TF-VIIa-Xa complex formation), or thrombin (5 nM) for 3 hours. Relative TF and CXCL8 mRNA abundance was determined by RT-PCR (n = 5-12 per condition). (C) EA.hy926 cells were pretreated with poly(I:C) (12.5 μg/mL) and thrombin (0.5-10 nM) for 6 hours, and the functionally active cell-surface TF was determined by an fXa-generation assay (n = 4). (D) Baseline abundance of TLR3, PAR1, and PAR2 mRNA relative to GAPDH mRNA in EA.hy926 cells and HUVECs. Target gene/GAPDH ratios in EA.hy926 cells were arbitrarily set to 1. (E) Changes in the abundance of PAR1, PAR2, and TLR3 mRNA relative to GAPDH mRNA in response to thrombin and/or poly(I:C) (n = 6) in EA.hy926 and HUVECs stimulated for 3 hours with poly(I:C) (12.5 μg/mL) and/or thrombin (5 nM). Baseline levels were arbitrarily set to 1. (F) TF and CXCL8 mRNA abundance in HUVECs and EA.hy926 cells treated for 3 hours with poly(I:C) (12.5 μg/mL) and/or thrombin (5 nM; n = 6/condition). All data represent the mean ± standard deviation of the indicated number of replicates and were generated in at least 2 biological replicates. The statistical significance of the differences between 2 groups was analyzed by Student t test. A comparison of more than 2 groups was conducted by ANOVA followed by a multiple-comparison test. *P < .05; **P < .01; ***P < .001; ****P < .0001. ns, nonsignificant.
EA.hy926 cells expressed higher TLR3, but lower Par1 mRNA levels than HUVECs (Figure 3D). Treatment with thrombin and/or poly(I:C) moderately reduced the expression of TLR3, Par1, and Par2 in HUVECs, whereas EC TLR3 was increased approximately twofold in EaHy.926 cells by poly(I:C) alone, but not by cotreatment with both agonists (Figure 3E). Correspondingly, HUVECs and EA.hy926 cells exhibited increased sensitivity to stimulation with thrombin or poly(I:C), respectively (Figure 3F).
Thrombin amplifies the poly(I:C) response via Par1-Par2 heterodimers
Thrombin activates Par1 via cleavage at Arg41, which may lead to nonproteolytic transactivation of Par2.53 Alternatively, thrombin bound to Thbd may directly cleave and activate Par2 at Arg36.54 To address the latter mechanism, we generated Thbd-deficient EA.hy926 cells via CRISPR/Cas9-mediated gene editing (Figure 4A). Thrombin augmented the abundance of TF- and CXCL8-mRNA in poly(I:C)-treated, Thbd-deficient cells to an extent similar to that in Thbd-expressing wild-type cells (Figure 4B), arguing against a significant role of direct Par2 cleavage by the thrombin-Thbd complex.
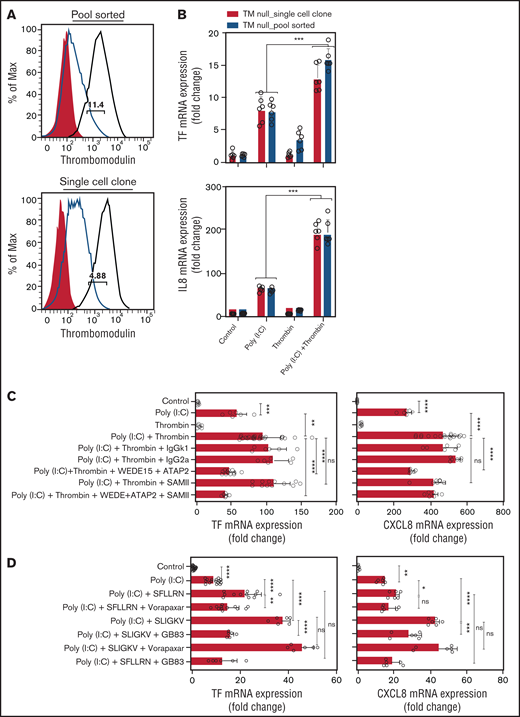
Thrombin augments poly(I:C)-induced TF and CXCL8 mRNA induction by activating PAR1/2 heterodimers. (A) Cell surface Thbd expression level in pool-sorted and single-cell clone Thbd-deficient EA.hy926 cells by flow cytometry. Shaded, nonimmune isotype control; black, parent (Thbd-expressing) cells; and red, Thbd-deficient cells. The numbers in the plots indicate the percentage of estimated maximum contamination, with cells expressing Thbd from 1 residual allele. (B) TM-deficient, pool-sorted cells and single-cell clones were stimulated with poly(I:C) (12.5 μg/mL) and thrombin (5 nM) for 3 hours. TF and CXCL8 mRNA abundance relative to GAPDH was measured by RT-PCR (n = 3-6; in duplicate). (C) EA.hy926 cells were pretreated with PAR1 (WEDE15, 10 μg/mL; ATAP2 10 μg/mL) and/or PAR2 (SAM-11, 10 μg/mL) cleavage-blocking antibodies, followed by treatment with poly(I:C) (12.5 μg/mL) and/or thrombin (5 nM) for 3 hours. TF and CXCL8 mRNA abundance relative to GAPDH was measured by RT-PCR (n = 3-12; in duplicate). (D) EA.hy926 cells were pretreated with PAR-specific inhibitors (PAR1, vorapaxar, 1 μM; PAR2, GB83, 25 μM) followed by stimulation with poly(I:C) (12.5 μg/mL) and PAR1/2 agonist peptides. TF and CXCL8 mRNA abundance relative to GAPDH mRNA was measured by RT-PCR (n = 3-9; in duplicate). Data are expressed as the mean ± standard deviation. The statistical significance of differences between groups was analyzed by ANOVA followed by the multiple-comparison test. *P < .05; **P < .01; ***P < .001; ****P < .0001. ns, nonsignificant.
Thrombin augments poly(I:C)-induced TF and CXCL8 mRNA induction by activating PAR1/2 heterodimers. (A) Cell surface Thbd expression level in pool-sorted and single-cell clone Thbd-deficient EA.hy926 cells by flow cytometry. Shaded, nonimmune isotype control; black, parent (Thbd-expressing) cells; and red, Thbd-deficient cells. The numbers in the plots indicate the percentage of estimated maximum contamination, with cells expressing Thbd from 1 residual allele. (B) TM-deficient, pool-sorted cells and single-cell clones were stimulated with poly(I:C) (12.5 μg/mL) and thrombin (5 nM) for 3 hours. TF and CXCL8 mRNA abundance relative to GAPDH was measured by RT-PCR (n = 3-6; in duplicate). (C) EA.hy926 cells were pretreated with PAR1 (WEDE15, 10 μg/mL; ATAP2 10 μg/mL) and/or PAR2 (SAM-11, 10 μg/mL) cleavage-blocking antibodies, followed by treatment with poly(I:C) (12.5 μg/mL) and/or thrombin (5 nM) for 3 hours. TF and CXCL8 mRNA abundance relative to GAPDH was measured by RT-PCR (n = 3-12; in duplicate). (D) EA.hy926 cells were pretreated with PAR-specific inhibitors (PAR1, vorapaxar, 1 μM; PAR2, GB83, 25 μM) followed by stimulation with poly(I:C) (12.5 μg/mL) and PAR1/2 agonist peptides. TF and CXCL8 mRNA abundance relative to GAPDH mRNA was measured by RT-PCR (n = 3-9; in duplicate). Data are expressed as the mean ± standard deviation. The statistical significance of differences between groups was analyzed by ANOVA followed by the multiple-comparison test. *P < .05; **P < .01; ***P < .001; ****P < .0001. ns, nonsignificant.
Antibodies ATAP2 and WEDE15, which block Arg41 cleavage and thrombin binding to PAR1,55,56 prevented thrombin-mediated augmentation of poly(I:C)-induced TF mRNA abundance. In contrast, the PAR2 cleavage-blocking antibody SAM-11 had no effect (Figure 4C). The PAR2 antagonist GB83 abrogated the increase in TF- and CXCL8 mRNA (Figure 4D), but did not inhibit the mobilization of intracellular Ca++ by thrombin or the PAR1-selective agonist peptide TFLLRN-NH2, indicating that the inhibitory effect of GB83 is unlikely to involve off-target effects on PAR1 (supplemental Figure 1). In contrast, the PAR1 antagonist vorapaxar had no effect on the response to the PAR2 agonist peptide (Figure 4D), which suggests that thrombin augmented TF and CXCL8 mRNA abundance in poly(I:C)-stimulated cells by cleaving PAR1 at Arg41 and most likely by nonproteolytic transactivation of PAR2 by the PAR1-tethered ligand.
Thrombin amplifies poly(I:C)-induced endothelial TF procoagulant function
Immunohistological detection with anti-TF antibodies in permeabilized, poly(I:C)-treated EA.hy926 cells revealed robust augmentation of TF antigen levels by Par1 and Par2 agonist peptides (Figure 5A) that was corroborated by western blot analysis of whole-cell lysates (Figure 5B), procoagulant activity in whole-cell extracts (Figure 5C), and measurement of cell surface–associated TF activity (Figure 5D). Akin to data obtained with agonist peptides, thrombin-induced TF antigen correlated with functional TF activity in whole-cell lysates (Figure 5E), with immunostaining of cell surface-associated TF in nonpermeabilized cells (Figure 5F), and with cell surface–associated TF activity, as measured by a 2-stage factor Xa generation assay (Figure 5G). As for TF mRNA, augmentation of whole-cell TF antigen (Figure 5H) and cell surface TF activity (Figure 5I) were sensitive to pharmacologic inhibition of both Par1 and Par2. Likewise, antibodies blocking cleavage of Par1 (ATAP2/WEDE15), but not Par2 (SAM-11), suppressed cell surface TF activity by thrombin/poly(I:C) costimulation (Figure 5I). The amplification of functional TF activity in costimulated cells therefore appeared to be regulated predominantly at the level of TF gene transcription and/or mRNA stability.
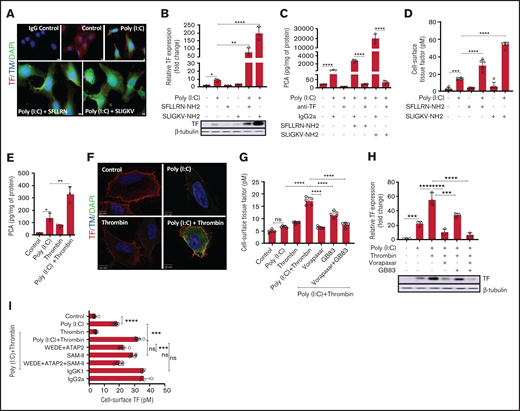
Thrombin-PAR1/2 signaling amplifies cell surface-associated TF procoagulant activity in poly(I:C)-treated cells. (A) EA.hy926 cells were stimulated with poly(I:C) (12.5 μg/mL) and PAR1-selective (SFLLR) or PAR2-selective agonist peptides (150 μM) for 4 hours, and TF and THBD antigen were detected by immunofluorescence in permeabilized cells. Original magnification, ×40; bar represents 10 μm; nuclear counterstain with 4′6-diamidino-2-phenylindole (n = 3). (B) EA.hy926 cells were stimulated with poly(I:C) (12.5 μg/ml) and PAR1 and PAR2 activation peptides for 6 hours in triplicate, and the protein levels of TF and β-tubulin were determined by western blot analysis with the respective antibodies. Data shown in the bar graph represent the mean ± standard deviation of the ratio of TF to β-tubulin, determined by quantitative densitometry of western blots (n = 3) . (C) Cell lysates were prepared from EA.hy926 cells stimulated with poly(I:C) (12.5 μg/mL) and PAR1- and PAR2-activation peptides (150 μM) for 6 hours. TF procoagulant activity (PCA) was measured in the presence or absence of anti-TF antibody (HTF-1; 10 µg/mL) or IgG control (IgG2a; 10 μg/mL) by a 1-stage clotting assay and converted into picograms TF per milligrams protein from a standard curve generated with recombinant TF (Innovin; n = 3). (D) Cell-surface TF activity on intact cells was determined by an fXa generation assay (n = 5-6). (E) EA.hy926 cells were stimulated for 6 hours with poly(I:C) and/or thrombin, and TF procoagulant activity was measured by a 1-stage clotting assay and converted into PCA via a standard curve generated with recombinant TF (Innovin; n = 3). (F) Immunofluorescence detection of cell-surface TF and Thbd antigen on EA.hy926 cells stimulated for 4 hours with poly(I:C) and/or thrombin (original magnification, ×100; bar represents 10 μm; n = 3). (G) EA.hy926 cells were pretreated for 45 minutes with inhibitors of PAR1 (vorapaxar, 1 μM) and/or PAR2 (GB83, 25 μM) followed by stimulation with poly(I:C) and/or thrombin for 6 hours. Cell-surface TF activity was determined by fXa generation (n = 8). (H) EA.hy926 cells were pretreated for 45 minutes with PAR-specific inhibitors (vorapaxar, 1 μM; GB83, 25 μM) followed by stimulation with poly(I:C) and/or thrombin for 6 hours. Abundance of TF and β-tubulin in whole-cell lysates were determined by western blot analysis (n = 3). (I) EA.hy926 cells were pretreated (45 minutes) with cleavage-blocking antibodies for PAR1 (WEDE15, 10 μg/mL; ATAP2, 10 μg/mL) and/or PAR2 (SAM-11, 10 μg/mL), or isotype-matched nonimmune IgG controls followed by stimulation with poly(I:C) and/or thrombin for 6 hours. Cell-surface TF activity was determined via FXa generation assay (n = 4-8). Statistical significance was determined by ANOVA followed by a multiple-comparison test. *P < .05; **P < .01; ***P < .001; ****P < .0001. ns, not significant.
Thrombin-PAR1/2 signaling amplifies cell surface-associated TF procoagulant activity in poly(I:C)-treated cells. (A) EA.hy926 cells were stimulated with poly(I:C) (12.5 μg/mL) and PAR1-selective (SFLLR) or PAR2-selective agonist peptides (150 μM) for 4 hours, and TF and THBD antigen were detected by immunofluorescence in permeabilized cells. Original magnification, ×40; bar represents 10 μm; nuclear counterstain with 4′6-diamidino-2-phenylindole (n = 3). (B) EA.hy926 cells were stimulated with poly(I:C) (12.5 μg/ml) and PAR1 and PAR2 activation peptides for 6 hours in triplicate, and the protein levels of TF and β-tubulin were determined by western blot analysis with the respective antibodies. Data shown in the bar graph represent the mean ± standard deviation of the ratio of TF to β-tubulin, determined by quantitative densitometry of western blots (n = 3) . (C) Cell lysates were prepared from EA.hy926 cells stimulated with poly(I:C) (12.5 μg/mL) and PAR1- and PAR2-activation peptides (150 μM) for 6 hours. TF procoagulant activity (PCA) was measured in the presence or absence of anti-TF antibody (HTF-1; 10 µg/mL) or IgG control (IgG2a; 10 μg/mL) by a 1-stage clotting assay and converted into picograms TF per milligrams protein from a standard curve generated with recombinant TF (Innovin; n = 3). (D) Cell-surface TF activity on intact cells was determined by an fXa generation assay (n = 5-6). (E) EA.hy926 cells were stimulated for 6 hours with poly(I:C) and/or thrombin, and TF procoagulant activity was measured by a 1-stage clotting assay and converted into PCA via a standard curve generated with recombinant TF (Innovin; n = 3). (F) Immunofluorescence detection of cell-surface TF and Thbd antigen on EA.hy926 cells stimulated for 4 hours with poly(I:C) and/or thrombin (original magnification, ×100; bar represents 10 μm; n = 3). (G) EA.hy926 cells were pretreated for 45 minutes with inhibitors of PAR1 (vorapaxar, 1 μM) and/or PAR2 (GB83, 25 μM) followed by stimulation with poly(I:C) and/or thrombin for 6 hours. Cell-surface TF activity was determined by fXa generation (n = 8). (H) EA.hy926 cells were pretreated for 45 minutes with PAR-specific inhibitors (vorapaxar, 1 μM; GB83, 25 μM) followed by stimulation with poly(I:C) and/or thrombin for 6 hours. Abundance of TF and β-tubulin in whole-cell lysates were determined by western blot analysis (n = 3). (I) EA.hy926 cells were pretreated (45 minutes) with cleavage-blocking antibodies for PAR1 (WEDE15, 10 μg/mL; ATAP2, 10 μg/mL) and/or PAR2 (SAM-11, 10 μg/mL), or isotype-matched nonimmune IgG controls followed by stimulation with poly(I:C) and/or thrombin for 6 hours. Cell-surface TF activity was determined via FXa generation assay (n = 4-8). Statistical significance was determined by ANOVA followed by a multiple-comparison test. *P < .05; **P < .01; ***P < .001; ****P < .0001. ns, not significant.
Endothelial-leukocyte adhesion in costimulated cells requires PAR1 and PAR2
Replicating earlier findings,57,58 independent verification of the gene expression data by quantitative RT-PCR confirmed that thrombin and poly(I:C) each increased VCAM1 and E-selectin mRNA abundance and exerted a synergistic effect when given together (Figure 6A). Poly(I:C) and thrombin individually induced VCAM1 surface expression in 13% to 17% of cells (Figure 6B). Thrombin elicited coexpression of VCAM1 and E-selectin in ∼5.7% of cells after 4 hours of stimulation. Costimulation with poly(I:C) and thrombin elicited coexpression of VCAM1 and E-selectin on ∼20% of all cells, reflecting a ∼13-fold increase compared with cells stimulated with poly(I:C) alone (Figure 6B). Adhesion of THP-1 cells to HUVEC monolayers was significantly increased by poly(I:C) (∼8-fold) and further enhanced by costimulation with thrombin (∼15-fold) relative to untreated cells (Figure 6C-D). Pharmacologic inhibition of PAR1 or PAR2 with vorapaxar and GB83, respectively, suppressed THP-1 adhesion similar to the effect of VCAM1- or E-selectin–blocking antibodies (Figure 6C-D).
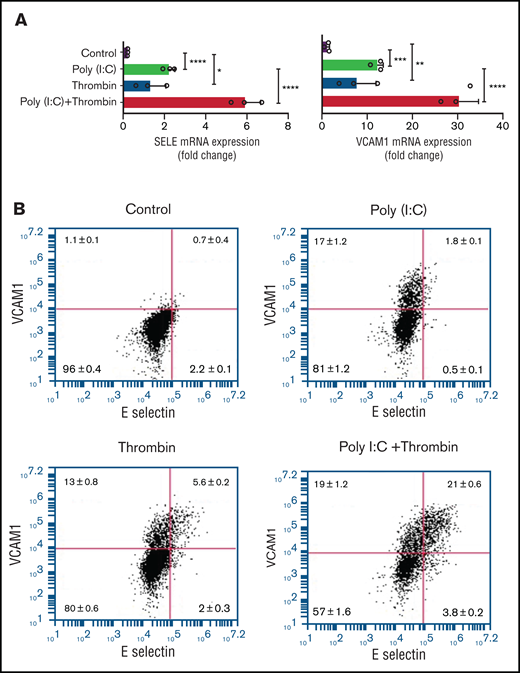
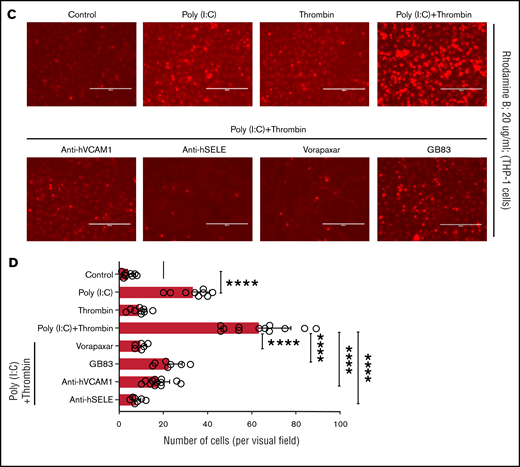
Leukocyte-endothelial interactions. (A) HUVECs were treated with poly(I:C) (12.5 μg/mL) and thrombin (5 nM) for 3 hours, and the abundance of E-selectin and VCAM-1 mRNA relative to GAPDH was measured by quantitative RT-PCR. (B) Representative scatterplots depicting cell surface expression of VCAM-1 and E-selection determined by flow cytometry in HUVECs treated for 4 hours with poly(I:C) and/or thrombin. (C-D) Representative micrographs of static adhesion assay of rhodamine B-labeled THP-1 cells to HUVECs treated with poly(I:C) and/or thrombin, vorapaxar, GB83, or anti-E-selectin– and anti-VCAM1 blocking antibodies (10 μg/mL) and respective nonimmune IgG controls (10 μg/mL). Original magnification, ×20. Brightness adjusted to 75% with Powerpoint picture correction. Bars represent 200 µm. (D) The mean ± standard deviation of adherent THP-1 cells per visual field from 2 independent experiments (n ≥ 9 for control, thrombin, poly(I:C); n ≥ 6 for anti-VCAM-1/E-selectin). *P < .05; **P < .01; ***P < .001; ****P < .0001 by ANOVA, followed by multiple comparison test.
Leukocyte-endothelial interactions. (A) HUVECs were treated with poly(I:C) (12.5 μg/mL) and thrombin (5 nM) for 3 hours, and the abundance of E-selectin and VCAM-1 mRNA relative to GAPDH was measured by quantitative RT-PCR. (B) Representative scatterplots depicting cell surface expression of VCAM-1 and E-selection determined by flow cytometry in HUVECs treated for 4 hours with poly(I:C) and/or thrombin. (C-D) Representative micrographs of static adhesion assay of rhodamine B-labeled THP-1 cells to HUVECs treated with poly(I:C) and/or thrombin, vorapaxar, GB83, or anti-E-selectin– and anti-VCAM1 blocking antibodies (10 μg/mL) and respective nonimmune IgG controls (10 μg/mL). Original magnification, ×20. Brightness adjusted to 75% with Powerpoint picture correction. Bars represent 200 µm. (D) The mean ± standard deviation of adherent THP-1 cells per visual field from 2 independent experiments (n ≥ 9 for control, thrombin, poly(I:C); n ≥ 6 for anti-VCAM-1/E-selectin). *P < .05; **P < .01; ***P < .001; ****P < .0001 by ANOVA, followed by multiple comparison test.
Discussion
In the current work, we investigated how 2 signals associated with viral infections (ie, the viral RNA analogue poly[I:C]) and enhanced generation of signaling-competent blood coagulation proteases interact to alter the function of vascular ECs. Interrogation of the HUVEC transcriptome by RNA profiling showed that costimulation with thrombin and poly(I:C) induced a state of inflammatory endothelial activation distinct from that elicited by either signal alone. This response reflected in part the cooperative effects of the individual agonists (34 down, 112 up; Figure 1; Venn diagram, E-G), but also included a large number of transcripts that were significantly regulated only in costimulated cells (120 down; 127 up; Venn diagram C). Pathway analysis of gene expression data indicated that the response of genes selectively upregulated in costimulated cells predominantly reflected transcriptional activation via NF-κB complexes, whereas the subset of transcripts selectively downregulated by co stimulation may have a stronger effect on the maintenance of vascular integrity.
Although the inflammatory effect of thrombin signaling in ECs is well documented,24,59-62 only a small fraction of transcripts (17 of 292; Figure 1, Venn section F vs sum of sections A,B,D,E,F,G) was upregulated individually by both thrombin or poly(I:C) under our experimental conditions. Yet, genes within this group exhibited overall the most robust, cooperative response to costimulation (Table 1) and included endothelial adhesion receptors and cytokines central to the regulation of leukocyte accumulation at sites of infection (E-selectin/SELE, VCAM1, CXCL8, IL-6, CXCL2, and CCL20). Similarly, whereas poly(I:C) alone, compared with thrombin, had only a modest effect on TF mRNA, it conveyed a very marked synergistic effect on TF mRNA abundance in costimulated cells. A key observation from the analyses was therefore that the coincident activation of TLR3 by poly(I:C) and of PAR1 and 2 by thrombin selectively elicited a synergistic activation of the 2 critical biological mechanisms promoting endothelial thromboinflammatory functions: the initiation of blood coagulation and the control of leukocyte trafficking.
The correlations between mRNA abundance, protein antigen levels, and surface expression, as well as functional activity for TF and the adhesion receptors VCAM1 and E-selectin, indicate that the proinflammatory and procoagulant effects of poly(I:C)/thrombin costimulation are regulated predominantly at the level of gene transcription and/or mRNA stability. However, thrombin or poly(I:C) each enhanced the abundance of VCAM1 and E-selectin mRNA, but only VCAM1 antigen expression was induced on the cell surface by either ligand alone. In contrast, costimulation with poly(I:C) and thrombin led to significant coexpression of both receptors, which is likely to be necessary for efficient leukocyte tethering.63
To gain a mechanistic understanding of the relative contributions of direct thrombin signaling and secondary TF-dependent signaling, we showed that the effect of thrombin could be replicated only by synthetic tethered ligands derived from PAR1 and PAR2, but not from PAR3 or PAR4. Of note, PAR2-selective agonists consistently yielded a more robust response than PAR1- or PAR1/2-elective agonists, indicating the dominant role of PAR2 activation. We investigated 3 established paradigms of PAR2 activation: (1) activation of PAR1/2 by fXa, the TF-VIIa complex, or the Tf-VIIa-Xa complex22,23; (2) direct activation of PAR2 by thrombin or the thrombin-Thbd complex54,64; and (3) transactivation of PAR2 by thrombin-cleaved PAR1.53 Although fXa has been reported to induce TF activity in HUVECs,65 we ruled out a significant role of a TF-dependent mechanism, direct signaling by factors Xa and VIIa generated as a consequence of TF-mediated coagulation initiation, and direct activation of PAR2 by thrombin-Thbd complexes. Instead, our observations indicate that thrombin is the principal physiologic protease triggering thromboinflammatory feedback amplification via proteolytic cleavage of PAR1, and potentially, transactivation of PAR2 by the PAR1-tethered ligand. Given that PAR2 inhibition completely blocked the effects of a PAR1 agonist, we suspect that the relevant thrombin target is indeed a physical PAR1/2 heterodimer, in which PAR1 inhibition with vorapaxar is still compatible with the agonist-induced transition of PAR2 into a signaling-competent conformation. Independent earlier studies showed that induction of decay-accelerating factor by thrombin likewise proceeds through activation of PAR1/2 heterodimers.66 Notably, in this study PAR1 activation was coupled to PKC-α, whereas PAR2 signaling required PKC-ε. Our own experimental approaches were not informative as to whether the cooperativity between PAR1 and PAR2 reflects intracellular integration of distinct signaling cascades initiated via each receptor or is caused by a specific signaling platform composed of physical PAR1/2 heterodimers. A further limitation in the interpretation of data from cross-inhibition experiments with PAR1/2 agonists and GB83 is the known cross-reactivity of the PAR1 agonist SFLLRN-NH2 with PAR2 and the albeit limited potential for PAR1 inhibition by the PAR2-selective inhibitor GB83.67 Our findings also suggest that selective, physiologically relevant agonists of PAR2 may elicit, or propagate a similar modulation of the poly(I:C) response, as shown herein for thrombin. For example, in the presence of fVII and fX, the initial poly(I:C)-induced increase in TF expression could lead to direct PAR2 activation by the TF-fVIIa and/or TF-fVIIa-Xa complexes, or indirectly via transactivation of promatriptase by the ternary TF-fVIIa-fXa complex and ensuing PAR2 activation by matriptase.68
A seminal report on the importance of PAR1/2 heterodimers in mouse models of endotoxemia and sterile inflammation described the disease stage–dependent opposing effects of PAR1 activation over the course of sepsis, with PAR1 activation being detrimental in the early stages, but protective in the later stages.69 In that study, results of in vitro experiments on human ECs suggested that the switch from detrimental to protective PAR1 effects requires PAR2 and involves the lipopolysaccharide (LPS)-induced recruitment of intracellular PAR2 into cell surface–associated PAR1/2 heterodimers. The study did not ascertain that the in vivo protective effects of PAR1/2 agonism were indeed dependent on endothelial PAR2 expression and used different inflammatory stimuli (LPS) and readouts of endothelial function (barrier integrity) than our current work. Nevertheless, these observations raise new questions of to what extent the apparent role of PAR1/2 heterodimers may depend on the species investigated, the specific nature of the inflammatory/infectious challenge (LPS-TLR4 vs poly[I:C]-TLR3), the effects on non-EC types, and the specific biased or unbiased mechanisms of action of PAR1/2 agonists.
The concept that inflammatory mediators alter the response capacity of ECs to thrombin has been demonstrated earlier,70 Preincubation of HUVECs with TNFα and LPS significantly inhibited, rather than amplified, thrombin-mediated induction of VCAM1, ICAM, TF, and E-selectin. This result strongly suggests that the endothelial thrombin response may vary substantially, depending on the specific mediator mix present in the blood of patients with a given disease.71,72 In addition, different types of ECs likely exhibit distinct cytokine responses in vitro,72 as well as organ-specific LPS responses in vivo.73
In summary, our findings expand the existing evidence that inflammatory cytokines and danger signals other than poly(I:C), as shown here, synergize with thrombin and other PAR agonists, such as matrix metalloprotease 1 and pepducins, to modify and amplify thromboinflammatory functions of the endothelium.59,69,74-77 The amplification of TF procoagulant activity by thrombin in poly(I:C)-treated cells implies that thrombin, whether initially generated as a consequence of TF expression on inflammatory immune cells, such as macrophages, or on infected endothelium itself, may sustain and amplify its own formation by further enhancing TF expression on endothelium. In essence, this mechanism therefore constitutes a feedback amplification of thromboinflammatory functions of virus-exposed endothelium. Mechanistically, the current work showed that this thrombin-mediated feedback requires PAR1/2 heterodimers and may be inhibited by PAR2 antagonists, providing an incentive for further development and evaluation of monobivalent or heterobivalent antagonists of PAR1-PAR2 heterodimers78 as a potential therapeutic to reduce endothelial dysfunction.
Acknowledgments
The authors thank Wolfram Ruf and Skip Brass for providing reagents and the BRI Core facility staff for their support and expertise.
This work was supported by National Institutes of Health, National Heart, Lung, and Blood Institute grants R01HL142799 (S.A.), R01HL119523 (N.M.), R01HL117132 (H.W.), and R15HL127636 (C.D.) and the Office of the Director R25OD014807 (C.F.); and by the Richard Gallagher Fellowship Award from the Versiti Blood Research Foundation (S.S.).
Authorship
Contribution: S.S. performed the experiments, analyzed the data, and contributed to writing the manuscript; Y.O., I.H., and F.B. performed the experiments; R.B., S.S., and H.W. analyzed the RNA-sequencing data; N.M., S.A., and C.F. provided expert technical advice and contributed to the study; M.Z. and I.H. conducted CRISPR/Cas9 genome editing and flow cytometry; and S.S. and H.W. designed the experiments, analyzed the data, and wrote the manuscript.
Conflict-of-interest disclosure: C.D. is the founder of a company (Function Therapeutics LLC) that is developing anti-inflammatory PAR ligands (not included in this manuscript). The remaining authors declare no competing financial interests.
The current affiliation for S.S. is Pulmonary Center, Boston University School of Medicine, Boston, MA.
Correspondence: Hartmut Weiler, Blood Research Institute, Versiti, 8727 Watertown Plank Rd, Milwaukee, WI 53226; e-mail: hweiler@versiti.org.

