

Key Points
Cytogenetic abnormalities differ significantly and confer distinct prognostic impact in each subgroup of CBF-AML.
Survival was improved for patients with co-occurrence of inv(16) with trisomy 8 or t(8;21) with hyperdiploidy, hypodiploidy, and del(9p).

Abstract
Patients with core-binding factor (CBF) acute myeloid leukemia (AML), caused by either t(8;21)(q22;q22) or inv(16)(p13q22)/t(16;16)(p13;q22), have higher complete remission rates and longer survival than patients with other subtypes of AML. However, ∼40% of patients relapse, and the literature suggests that patients with inv(16) fare differently from those with t(8;21). We retrospectively analyzed 537 patients with CBF-AML, focusing on additional cytogenetic aberrations to examine their impact on clinical outcomes. Trisomies of chromosomes 8, 21, or 22 were significantly more common in patients with inv(16)/t(16;16): 16% vs 7%, 6% vs 0%, and 17% vs 0%, respectively. In contrast, del(9q) and loss of a sex chromosome were more frequent in patients with t(8;21): 15% vs 0.4% for del(9q), 37% vs 0% for loss of X in females, and 44% vs 5% for loss of Y in males. Hyperdiploidy was more frequent in patients with inv(16) (25% vs 9%, whereas hypodiploidy was more frequent in patients with t(8;21) (37% vs 3%. In multivariable analyses (adjusted for age, white blood counts at diagnosis, and KIT mutation status), trisomy 8 was associated with improved overall survival (OS) in inv(16), whereas the presence of other chromosomal abnormalities (not trisomy 8) was associated with decreased OS. In patients with t(8;21), hypodiploidy was associated with improved disease-free survival; hyperdiploidy and del(9q) were associated with improved OS. KIT mutation (either positive or not tested, compared with negative) conferred poor prognoses in univariate analysis only in patients with t(8;21).
Introduction
Acute myeloid leukemia (AML) is one of the most common hematologic malignancies. It is primarily a disease of older adults, with a median age of 69 years in the United States.1 Although up to 75% of patients with AML achieve complete remission (CR) after induction chemotherapy, the majority of patients achieving CR eventually relapse (54% to 92%, depending on cytogenetic risk).2
Core-binding factor (CBF) AML is a subgroup of AML characterized by distinct cytogenetic abnormalities, molecular pathogenesis, and a favorable prognosis. CBF is a DNA-binding heterodimeric transcription factor comprising α- and β-subunits encoded by the RUNX1 and CBFB genes, respectively. CBF regulates multiple hematopoietic genes responsible for myeloid differentiation.3-5 Chromosomal aberrations t(8;21)(q22;q22) and inv(16)(p13q22) or t(16;16)(p13;q22), hereafter abbreviated as t(8;21) and inv(16), create, respectively, the RUNX1-RUNX1T1 and CBFB-MYH11 gene fusions. The RUNX-1-RUNX-1T1 chimeric protein contributes to leukemogenesis primarily by disrupting normal hematopoiesis via its constitutive transcriptional activity, which increases the capacity of hematopoietic precursors for self-renewal and decelerates their terminal differentiation.6 CBFB-MYH-11 disrupts hematopoiesis because it associates with corepressor complexes, resulting in recruitment of histone deacetylase activity and silencing of gene function, and sequesters RUNX-1 protein in the cytoplasm.6
AML with these CBF gene fusions (CBF-AML), defined by the aforementioned cytogenetic abnormalities, comprise 12% to 15% of adult AML.7,8 CBF-AML is clinically characterized by relatively high complete remission (CR) rates and long survival; thus, its prognosis is considered “favorable” compared with that of other subtypes of AML.9 Both the National Comprehensive Cancer Network10 and the European LeukemiaNet7 do not recommend allogeneic hematopoietic cell transplantation (alloHCT) during the first complete remission (CR1), with a possible exception of patients with t(8;21) and KIT mutations. Given the similarity in genetic background and prognosis, CBF-AML with inv(16) and t(8;21) are often considered to be a single disease entity and are treated similarly. However, up to 40% of patients with CBF-AML die of treatment failure, mainly from recurrence.11,12 Moreover, many studies suggest that CBF-AML with inv(16) differs from CBF-AML with t(8;21) in demographic, cytogenetic, molecular, and clinical features.6,7,13-15 Better genomic characterization of CBF-AML could aid in identifying different prognostic and therapeutic groups to improve outcomes for these patients. Minimal or measurable residual disease recommendations for monitoring of RUNX1-RUNX1T1 and CBFB-MYH11 transcripts are outlined in the 2017 European LeukemiaNet guidelines7 and are important in the clinical management of patients with AML.16
In this retrospective study, we analyzed additional chromosomal abnormalities in 537 patients with CBF-AML, to our knowledge the largest cytogenetic study in this relatively rare AML subgroup, to further refine differences between inv(16) and t(8;21) in treatment response, clinical outcomes, and associated molecular abnormalities.
Patients and methods
Data on 537 patients with CBF-AML were collected collaboratively from 12 institutions in the United States and Europe. There were 290 patients with inv(16) and 247 patients with t(8;21) with clinical and laboratory characteristics described in Table 1 and Figure 1. All patients were diagnosed from July 1996 through January 2017 and were required to have a bone marrow biopsy at diagnosis and after induction therapy at the pathology department of participating institutions. The presence of CBF-AML rearrangements, t(8;21)(q22;q22), inv(16)(p13q22), or t(16;16)(p13;q22), was assessed by conventional karyotyping, fluorescence in situ hybridization, and/or reverse transcription-polymerase chain reaction, for detection of RUNX1-RUNX1T1 and CBFB-MYH11 at each reporting institution. When available, conventional karyotyping data were captured at diagnosis. To be included in the subclone and secondary abnormalities analysis, a minimum of 2 metaphases was required for samples with limited karyotype studies (Figure 1). Analyses of NPM1, FLT3, and KIT mutations, including KIT D816V mutations, were performed at the individual institutions. Data were uniformly collected by completing a predesigned data spreadsheet at each institution. Patient identifiers were removed, and cases coded before data transfer to the University of Minnesota, where the main database was created and managed. The study was approved by the Institutional Review Board Human Subjects Committee at the University of Minnesota and was conducted in accordance with the Declaration of Helsinki.
Patient characteristics
| Characteristic | inv(16) (n = 290) | t(8;21) (n = 247) | Total (N = 537) | P |
|---|---|---|---|---|
| Age at diagnosis, y | .14 | |||
| Median (SD) | 50 (17) | 47 (18) | 48 (17) | |
| Range | 5-81 | 2-81 | 2-81 | |
| Sex, n (%) | .20 | |||
| Female | 134 (49) | 101 (44) | 235 (47) | |
| Male | 138 (51) | 131 (56) | 269 (53) | |
| Race, n (%) | .11 | |||
| White | 219 (79) | 171 (73) | 390 (76) | |
| Non-White | 60 (21) | 65 (27) | 125 (24) | |
| Type of AML, n (%) | .14 | |||
| De novo | 240 (87) | 194 (82) | 434 (84) | |
| Secondary | 37 (13) | 43 (18) | 80 (16) | |
| Year of diagnosis | .02 | |||
| 1995-1999 | 11 (4) | 14 (6) | 25 (5) | |
| 2000-2005 | 58 (21) | 29 (12) | 87 (17) | |
| 2006-2010 | 98 (35) | 105 (44) | 203 (39) | |
| 2011-2016 | 114 (40) | 89 (38) | 203 (39) | |
| WBC at diagnosis, ×109/L | <.01 | |||
| Median (SD) | 20 (57) | 11 (19) | 14 (46) | |
| Range | 1-373 | 1-140 | 1-373 | |
| Platelet at diagnosis, ×109/L | .49 | |||
| Median (SD) | 34 (78) | 32 (88) | 33 (82) | |
| Range | 1-646 | 6-445 | 1-646 |
| Characteristic | inv(16) (n = 290) | t(8;21) (n = 247) | Total (N = 537) | P |
|---|---|---|---|---|
| Age at diagnosis, y | .14 | |||
| Median (SD) | 50 (17) | 47 (18) | 48 (17) | |
| Range | 5-81 | 2-81 | 2-81 | |
| Sex, n (%) | .20 | |||
| Female | 134 (49) | 101 (44) | 235 (47) | |
| Male | 138 (51) | 131 (56) | 269 (53) | |
| Race, n (%) | .11 | |||
| White | 219 (79) | 171 (73) | 390 (76) | |
| Non-White | 60 (21) | 65 (27) | 125 (24) | |
| Type of AML, n (%) | .14 | |||
| De novo | 240 (87) | 194 (82) | 434 (84) | |
| Secondary | 37 (13) | 43 (18) | 80 (16) | |
| Year of diagnosis | .02 | |||
| 1995-1999 | 11 (4) | 14 (6) | 25 (5) | |
| 2000-2005 | 58 (21) | 29 (12) | 87 (17) | |
| 2006-2010 | 98 (35) | 105 (44) | 203 (39) | |
| 2011-2016 | 114 (40) | 89 (38) | 203 (39) | |
| WBC at diagnosis, ×109/L | <.01 | |||
| Median (SD) | 20 (57) | 11 (19) | 14 (46) | |
| Range | 1-373 | 1-140 | 1-373 | |
| Platelet at diagnosis, ×109/L | .49 | |||
| Median (SD) | 34 (78) | 32 (88) | 33 (82) | |
| Range | 1-646 | 6-445 | 1-646 |
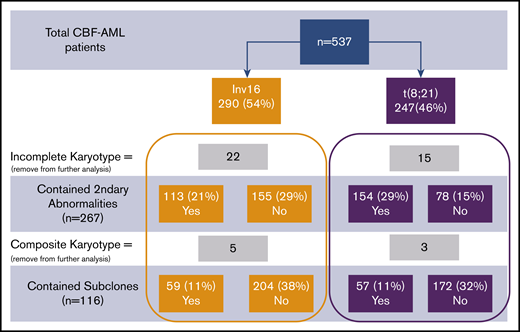
Consort diagram of patient cohort. Percentages provided were calculated in comparison with total patients.
Consort diagram of patient cohort. Percentages provided were calculated in comparison with total patients.
Definitions
A complex karyotype was defined as the presence of 3 or more chromosome abnormalities (ie, t(8;21) or inv(16) and 2 or more secondary abnormalities) detected by standard cytogenetic analysis. The presence of a subclone or subclones was recorded if there were 2 or more abnormal, cytogenetically distinct clones detected by conventional G banding. The dominant clone was defined as the one with the highest number of metaphases; the remaining clones were characterized as subclones. The presence of cells with a normal karyotype was not counted as a subclone but was analyzed separately. Secondary cytogenetic abnormalities were defined as those occurring in addition to either inv(16) or t(8;21) by conventional karyotype. Each specific secondary cytogenetic abnormality was counted only once, even if the same secondary abnormality was present in 2 or more related clones. Secondary abnormalities known to be recurrent in CBF-AML (ie, trisomy of chromosomes 4, 8, 13, 21, and 22; deletion of 9q; and loss of the X or Y chromosome) were counted, even in those instances when they were present in only 1 metaphase. Other cytogenetic abnormalities, which are defined as any cytogenetic abnormalities except those aforementioned secondary abnormalities recurrent in CBF-AML, were counted only when they were clonal (that is, a structural abnormality or a trisomy detected in at least 2 metaphases and a monosomy found in at least 3 metaphases. Categories of secondary abnormalities designated “del(7q)” and “del(9q)” contain mostly deletions of 7q and 9q, respectively, but they also include relatively rare instances of monosomy of chromosomes 7 and 9, as well as other structural abnormalities that result in loss of genetic material from 7q and 9q (eg, add(7q) or add(9q)). Loss of chromosome X or Y categories included only whole chromosome losses of these sex chromosomes. Loss of the X chromosome was counted only in female patients and loss of the chromosome Y only in male patients. Pseudodiploidy was defined as abnormal karyotypes with inv(16) or t(8;21) and a total of 46 chromosomes in all abnormal clones. If the total number of chromosomes was higher or lower than 46 chromosomes in any clone, then the ploidy level was defined as hyperdiploid or hypodiploid, respectively. All patients’ karyotypes were reevaluated and defined by a cytogeneticist (K.M.) for the study.
Statistical analysis
Statistical analysis focused on 2 general research objectives: to compare characteristics and outcomes of patients with inv(16) and t(8;21) and to analyze risk factors for outcomes separately within the inv(16) and t(8;21) groups. Patient characteristics were compared using standard descriptive statistics, the Wilcoxon rank sum test for continuous variables, and the χ2 test for categorical variables. Variables were prespecified and included those reported in Tables 1 and 2, plus those described in “Molecular abnormalities” and “Treatment” in “Results.” Disease-free survival (DFS) was defined as the time from achievement of CR to the time of relapse or death in remission, whichever occurred first. Subjects who survived without relapse were censored at last follow-up. Overall survival (OS) was defined as the time from diagnosis to the time of death from any cause. As with DFS, patients who survived were censored at last follow-up. The between-group differences in DFS and OS were described using Kaplan-Meier curves and compared by log-rank tests. Univariate Cox regression analyses were performed separately on DFS and OS for patients with inv(16) and t(8;21), so 4 models were fit per variable.
Cytogenetic characteristics of patients with inv(16) and those with t(8;21)
| Characteristic | inv(16) (n = 290) | t(8;21) (n = 247) | Total (N = 537) | P |
|---|---|---|---|---|
| Presence of subclones | .52 | |||
| No | 204 (78) | 172 (75) | 376 (76) | |
| Yes | 59 (22) | 57 (25) | 118 (24) | |
| Presence of normal metaphases | .52 | |||
| No | 153 (55) | 124 (52) | 277 (57) | |
| Yes | 108 (45) | 100 (48) | 208 (43) | |
| Presence of secondary cytogenetic abnormality(s) | <.01 | |||
| No | 155 (58) | 78 (34) | 233 (47) | |
| Yes | 113 (42) | 154 (66) | 267 (53) | |
| Deletion of 7q | .17 | |||
| No | 250 (96) | 209 (93) | 459 (94) | |
| Yes | 11 (4) | 16 (7) | 27 (6) | |
| Trisomy 8 | <.01 | |||
| No | 220 (84) | 209 (93) | 529 (88) | |
| Yes | 41 (16) | 16 (7) | 57 (12) | |
| Deletion of 9q | <.01 | |||
| No | 260 (99.6) | 192 (85) | 452 (93) | |
| Yes | 1 (0.4) | 33 (15) | 34 (7) | |
| Trisomy 13 | .09 | |||
| No | 255 (98) | 224 (99.6) | 479 (99) | |
| Yes | 6 (2) | 1 (0.4) | 7 (1) | |
| Trisomy 21 | <.01 | |||
| No | 245 (94) | 225 (100) | 470 (97) | |
| Yes | 16 (6) | 0 (0) | 16 (3) | |
| Trisomy 22 | <.01 | |||
| No | 217 (83) | 225 (100) | 442 (91) | |
| Yes | 44 (17) | 0 (0) | 44 (9) | |
| Loss of X in females | <.01 | |||
| No | 123 (100) | 57 (63) | 180 (85) | |
| Yes | 0 (0) | 33 (37) | 33 (15) | |
| Loss of Y in males | <.01 | |||
| No | 120 (95) | 71 (56) | 191 (75) | |
| Yes | 7 (5) | 55 (44) | 62 (25) | |
| Other secondary abnormalities | .30 | |||
| No | 216 (83) | 177 (79) | 393 (81) | |
| Yes | 45 (17) | 47 (21) | 92 (19) | |
| Complex karyotype | .02 | |||
| No | 218 (84) | 169 (75) | 387 (80) | |
| Yes | 42 (16) | 55 (25) | 97 (20) | |
| Ploidy level | <.01 | |||
| Hypodiploidy | 7 (3) | 87 (37) | 94 (18) | |
| Pseudodiploidy | 197 (72) | 126 (54) | 323 (64) | |
| Hyperdiploidy | 68 (25) | 22 (9) | 90 (18) |
| Characteristic | inv(16) (n = 290) | t(8;21) (n = 247) | Total (N = 537) | P |
|---|---|---|---|---|
| Presence of subclones | .52 | |||
| No | 204 (78) | 172 (75) | 376 (76) | |
| Yes | 59 (22) | 57 (25) | 118 (24) | |
| Presence of normal metaphases | .52 | |||
| No | 153 (55) | 124 (52) | 277 (57) | |
| Yes | 108 (45) | 100 (48) | 208 (43) | |
| Presence of secondary cytogenetic abnormality(s) | <.01 | |||
| No | 155 (58) | 78 (34) | 233 (47) | |
| Yes | 113 (42) | 154 (66) | 267 (53) | |
| Deletion of 7q | .17 | |||
| No | 250 (96) | 209 (93) | 459 (94) | |
| Yes | 11 (4) | 16 (7) | 27 (6) | |
| Trisomy 8 | <.01 | |||
| No | 220 (84) | 209 (93) | 529 (88) | |
| Yes | 41 (16) | 16 (7) | 57 (12) | |
| Deletion of 9q | <.01 | |||
| No | 260 (99.6) | 192 (85) | 452 (93) | |
| Yes | 1 (0.4) | 33 (15) | 34 (7) | |
| Trisomy 13 | .09 | |||
| No | 255 (98) | 224 (99.6) | 479 (99) | |
| Yes | 6 (2) | 1 (0.4) | 7 (1) | |
| Trisomy 21 | <.01 | |||
| No | 245 (94) | 225 (100) | 470 (97) | |
| Yes | 16 (6) | 0 (0) | 16 (3) | |
| Trisomy 22 | <.01 | |||
| No | 217 (83) | 225 (100) | 442 (91) | |
| Yes | 44 (17) | 0 (0) | 44 (9) | |
| Loss of X in females | <.01 | |||
| No | 123 (100) | 57 (63) | 180 (85) | |
| Yes | 0 (0) | 33 (37) | 33 (15) | |
| Loss of Y in males | <.01 | |||
| No | 120 (95) | 71 (56) | 191 (75) | |
| Yes | 7 (5) | 55 (44) | 62 (25) | |
| Other secondary abnormalities | .30 | |||
| No | 216 (83) | 177 (79) | 393 (81) | |
| Yes | 45 (17) | 47 (21) | 92 (19) | |
| Complex karyotype | .02 | |||
| No | 218 (84) | 169 (75) | 387 (80) | |
| Yes | 42 (16) | 55 (25) | 97 (20) | |
| Ploidy level | <.01 | |||
| Hypodiploidy | 7 (3) | 87 (37) | 94 (18) | |
| Pseudodiploidy | 197 (72) | 126 (54) | 323 (64) | |
| Hyperdiploidy | 68 (25) | 22 (9) | 90 (18) |
Data are expressed as the number of patients affected (percentage of study subgroups and total). Bold P values are statistically significant.
Variables of interest were recurrent additional cytogenetic abnormalities associated with CBF-AML (trisomy of chromosomes 4, 8, 13, 21, and 22, del(7q), del(9q); loss of chromosome X in females (in combination with t(8;21) only) or Y in males; complex karyotype; other abnormalities; and ploidy levels. A univariate model was fit for each variable of interest, which included prespecified factors for age, white blood cell (WBC) count at diagnosis, and KIT mutation status. To ensure that outliers of age and incomplete treatment data did not confound the study results, separate univariate analyses were performed on patients grouped by age range 15 to 65 years and compared with all patients. Similarly, a univariate analysis was also performed in patients whose complete chemotherapy data were available vs patients with incomplete chemotherapy data. Missing values for KIT mutation status were coded as the third-level “unknown.” Missing values on the other variables were removed from the statistical analysis. A sensitivity analysis was performed to determine the appropriateness of removing missing values if a proportion of missing values was large (a sensitivity test result is not included). Including all variables in a single multivariable model was not feasible because of the small number of events. We chose variables of interest with P < 0.1 from univariate analysis to include in the multivariate Cox regression, as well as age, WBC, and KIT mutation. A subset of these analyses is reported in Tables 3 and 4. The analyses were performed with SAS 9.4 (SAS Institutes, Cary, NC) and R 3.6.2.
MVA of survival outcomes in patients with inv(16)
| Covariate | HR | 95% CI | P |
|---|---|---|---|
| inv(16) DFS | |||
| Hypodiploidy vs pseudodiploidy | 0.532 | 0.122-2.325 | .402 |
| Hyperdiploidy vs pseudodiploidy | 0.681 | 0.339-1.370 | .281 |
| Trisomy 8 | 0.686 | 0.335-1.406 | .303 |
| Presence of secondary chromosomal abnormalities | 0.916 | 0.520-1.613 | .762 |
| inv(16) OS | |||
| Trisomy 8 | 0.397 | 0.184-.854 | .018 |
| Other chromosomal abnormalities | 2.052 | 1.186-3.550 | .010 |
| Covariate | HR | 95% CI | P |
|---|---|---|---|
| inv(16) DFS | |||
| Hypodiploidy vs pseudodiploidy | 0.532 | 0.122-2.325 | .402 |
| Hyperdiploidy vs pseudodiploidy | 0.681 | 0.339-1.370 | .281 |
| Trisomy 8 | 0.686 | 0.335-1.406 | .303 |
| Presence of secondary chromosomal abnormalities | 0.916 | 0.520-1.613 | .762 |
| inv(16) OS | |||
| Trisomy 8 | 0.397 | 0.184-.854 | .018 |
| Other chromosomal abnormalities | 2.052 | 1.186-3.550 | .010 |
All MVAs were adjusted for age, WBC count at diagnosis, and KIT mutation (positive, negative, and not tested).
MVA of survival outcomes in patients with t(8;21)
| Covariate | HR | 95% CI | P |
|---|---|---|---|
| t(8;21) DFS | |||
| Hypodiploidy vs pseudodiploidy | 0.414 | 0.210-0.815 | .011 |
| Hyperdiploidy vs pseudodiploidy | 0.386 | 0.139-1.073 | .068 |
| Deletion of 9q | 0.451 | 0.200-1.014 | .054 |
| Presence of secondary chromosomal abnormalities | 1.053 | 0.541-2.050 | .879 |
| t(8;21) OS | |||
| Hypodiploidy vs pseudodiploidy | 0.482 | 0.216-1.074 | .074 |
| Hyperdiploidy vs pseudodiploidy | 0.264 | 0.070-0.991 | .049 |
| Deletion of 7q | 1.516 | 0.589-3.899 | .388 |
| Deletion of 9q | 0.367 | 0.143-0.942 | .037 |
| Presence of secondary chromosomal abnormalities | 0.943 | 0.410-2.143 | .888 |
| Covariate | HR | 95% CI | P |
|---|---|---|---|
| t(8;21) DFS | |||
| Hypodiploidy vs pseudodiploidy | 0.414 | 0.210-0.815 | .011 |
| Hyperdiploidy vs pseudodiploidy | 0.386 | 0.139-1.073 | .068 |
| Deletion of 9q | 0.451 | 0.200-1.014 | .054 |
| Presence of secondary chromosomal abnormalities | 1.053 | 0.541-2.050 | .879 |
| t(8;21) OS | |||
| Hypodiploidy vs pseudodiploidy | 0.482 | 0.216-1.074 | .074 |
| Hyperdiploidy vs pseudodiploidy | 0.264 | 0.070-0.991 | .049 |
| Deletion of 7q | 1.516 | 0.589-3.899 | .388 |
| Deletion of 9q | 0.367 | 0.143-0.942 | .037 |
| Presence of secondary chromosomal abnormalities | 0.943 | 0.410-2.143 | .888 |
All MVAs were adjusted for age, WBC count at diagnosis, and KIT mutation (positive, negative, and not tested).
Results
Patient characteristics
Pretreatment patient characteristics are summarized in Table 1. The median follow-up time was 3.3 years (range, 0-17) for the inv(16) group and 2.5 years (range, 0-20) for the t(8;21) group. Baseline patient characteristics between the t(8;21) vs inv(16) groups were similar for age at diagnosis, sex, self-reported race, and secondary AML. However, the WBCs at diagnosis were significantly higher in inv(16)-bearing patients compared with t(8;21)-bearing patients (median, 20 vs 11 × 109/L; P < .01).
Secondary cytogenetic abnormalities and ploidy level
Thirty-seven patients did not have complete karyotype data: these patients were not included in the analysis of secondary abnormalities and subclones. Eight additional patients who had complex composite karyotypes were specifically removed from further analysis for subclones but were used in the analysis for secondary abnormalities. The presence of 1 or more cytogenetic subclones was found in 59 patients with inv(16) and in 57 patients with t(8;21) (P = .52; Figure 1). Secondary cytogenetic abnormalities occurred less frequently in patients with inv(16), 42% (n = 113), in comparison with 66% (n = 154) of patients with t(8;21).
We also examined specific secondary abnormalities. Patients with inv(16) more often harbored trisomy 8 than those with t(8;21) (16% vs 7%; P < .01). Trisomy 21 and 22 were detected in, respectively, 6% and 17% of patients with inv(16), but were not found in any of the patients with t(8;21) (P < .01 for both comparisons). In contrast, del(9q) (15% vs 0.4%; P < .01), loss of the X chromosome in females (37% vs 0%; P < .01), and loss of the Y chromosome in males (44% vs 5%; P < .01) were more frequent in t(8;21) than in those with inv(16). Other chromosomal abnormalities, except the aforementioned ones, were present in 17% of inv(16) and 21% of t(8;21) patients. Complex karyotypes were less frequent in patients with inv(16) than those with t(8;21) (16% vs 25%, P = .02). Some secondary abnormalities were relatively uncommon in CBF-AML and occurred with similar frequencies in patients with inv(16) and those with t(8;21). These include trisomy 4 detected in 1% of those with inv(16) and 2% of those with t(8;21), del(7q) found in 4% with inv(16) and 7% with t(8;21), and trisomy 13 observed in 2% with inv(16) and 0.4% with t(8;21). Separate comparisons of cytogenetic abnormalities and the presence or absence of cytogenetically normal cells were performed for secondary CBF-AML (secondary vs de novo) and age (65 < vs > 65 years), and did not show any significant differences, with the exception of the presence of cytogenetically normal cells being more frequent in both secondary CBF-AML (P = 0.01) and in patients older than 65 years (P = .05).
Regarding ploidy level, pseudodiploidy was most frequent in both cytogenetic categories of CBF-AML (72% in inv(16) and 54% in t(8;21) patients). Among patients with nonpseudodiploidy, hyperdiploidy was more frequent in those with inv(16) (25% vs 9%) whereas hypodiploidy was more common in those with t(8;21) (37% vs 3%; P < .01). A comparison of cytogenetic characteristics of patients with inv(16) and t(8;21) is shown in Table 2.
Molecular abnormalities
The mutation status of NPM1 and FLT3 was available for a small subset of patients in each cytogenetic group (15% with inv(16) and 11% with t(8;21)). Among patients with results available, NPM1 mutations were rare in patients with inv(16) (2 of 40; 5%), and absent in all 28 tested patients who had t(8;21). FLT3 mutations were also infrequent in patients with inv(16) (6 of 47; 13%) but were found in one-quarter of those with t(8;21) (10 of 39; 26%).
KIT mutation data were available for 339 patients. KIT mutations were found in 39 patients (13%) with inv(16) and in 41 patients (17%) with t(8;21) (P = .49). The most common type of KIT mutation, D816V, was detected in 31 patients (11%) with inv(16) and in 28 (11%) with t(8;21) (P = .66). Twenty-one patients showed KIT mutations that were not reported to be KIT D816V, but specific variant data were not captured for this database. One-hundred two patients (36%) with inv(16) and 85 (35%) with t(8;21) were not tested for KIT mutation. Among them, 50 patients (48%) with inv(16) and 32 (37%) with t(8;21) were diagnosed before 2006 when KIT mutations were first recognized as recurring in CBF-AML17,18 and routine testing for them was recommended.
Treatment
Among patients for whom treatment details were known, 180 of 209 (86%) of those with inv(16) and 131 of 168 (78%) of those with t(8;21), had 1 cycle of induction chemotherapy, and CR was achieved in 93% of those with inv(16) and 92% of those with t(8;21). Relapse occurred in 107 (37%) patients with inv(16) at a median of 11.9 months (range, 0.8-72.3) and in 60 (24%) patients with t(8;21) at a median of 10.6 months (range, 0.2-64.7 months). Eight patients with inv(16) AML and 5 with t(8;21) had an autologous hematopoietic cell transplantation. Ninety-seven patients (33%) with inv(16) AML underwent alloHCT with the following disease statuses: 28 with CR1 (10%), 59 with 2nd complete remission (CR2) (20%), 7 with active leukemia (2%), and 3 with unknown status (1%). AlloHCT was performed in 60 (24%) patients with t(8;21) AML with the following disease statuses: 31 with CR1 (13%), 19 with ≥CR2 (8%), and 10 with active leukemia (4%). Fifty-nine patients (61%) with inv(16) underwent alloHCT in CR2 or 3rd complete remssion compared with 19 patients (32%) with t(8;21) (P < .01). The analysis was repeated separating patients who had complete vs incomplete treatment data and no significant differences in OS or DFS. An analysis to evaluate whether there were treatment differences between intuitions showed that, of the 227 patients representing 6 institutions who had partial or complete chemotherapy information, the majority (82%) received at least 1 cycle of intensive chemotherapy at induction and/or consolidation. One institution used less intensive chemotherapy more often, and the overall numbers were low (n = 22) and not adequately powered for additional statistical analysis.
Survival
OS was similar in the 2 cytogenetic groups (P = .11): OS rate at 5 years was 68% (95% CI, 61-73) in patients with inv(16) and 62% (95% CI, 55-69) in those with t(8;21) (Figure 2A). The DFS rate at 5 years was significantly higher in patients with t(8;21), with 57% (95% CI, 50-65; P = .03) vs 47% (95% CI, 40-53) in the inv(16) group (Figure 2B). Of note, the survival of patients with missing chemotherapy data (ie, with either no treatment data available or with data on induction or consolidation therapy missing) was not different from survival of those with chemotherapy data available (as shown in supplemental Figure 1A for OS and supplemental Figure 1B for DFS). We also performed survival analyses stratified by complex cytogenetics defined as 4 or more cytogenetic abnormalities, with nonsignificant results for both DFS (P = .52) and OS (P = .42).

OS and DFS statistics in patients carrying inv(16) vs t(8;21). OS rates (A) were similar between the 2 groups at 5 years, and the DFS rate (B) at 5 years was significantly higher in patients with t(8;21).
OS and DFS statistics in patients carrying inv(16) vs t(8;21). OS rates (A) were similar between the 2 groups at 5 years, and the DFS rate (B) at 5 years was significantly higher in patients with t(8;21).
Impact of cytogenetic abnormalities and impact of the presence of subclones on survival
Univariable analysis for DFS and OS was performed and adjusted for 3 preselected variables mentioned in “Patients and methods” (results are shown in supplemental Tables 1-4). Hyperdiploidy, trisomy 8, and the presence of secondary chromosomal abnormalities were significant for DFS in the inv(16) group. Hypodiploidy or hyperdiploidy, del(9q), and the presence of secondary chromosomal abnormalities were associated with improved DFS and OS in patients with t(8;21); in contrast, del(7q) was associated with decreased OS. Notably, the presence of subclones did not affect the survival of patients in either cytogenetic group significantly. Univariate analysis comparing all patients to patients in an age range from 15 to 65 years, showed no significant difference for any variables including subclones or secondary abnormalities.
We performed multivariable analysis (MVA) adjusted for preselected age, WBC at diagnosis, and KIT mutation status (both positive and not tested). Chromosomal abnormalities other than trisomy 8 were associated with significantly inferior OS (hazards ratios [HR], 2.05; P = .01), whereas trisomy 8 was associated with superior OS (HR, 0.40; P = .02) in patients with inv(16) (Table 3). Del(9q) in patients with t(8;21) was associated with significantly improved OS (HR, 0.37; P = .04). Hypodiploidy was associated with significantly superior DFS (HR, 0.41; P = .01), whereas hyperdiploidy was associated with significantly better OS (HR, 0.26; P = .05) in patients with t(8;21) (Table 4).
Discussion
Using the largest cytogenetic data set of cases of CBF-AML, we characterized the clinical impact of additional cytogenetic abnormalities in patients with CBF-AML, defined by either inv(16) or t(8;21), and the results revealed specific prognostic patterns in the 2 groups.17,19 Certain cytogenetic abnormalities were more prevalent in 1 of the 2 CBF-AML entities: trisomy 22, trisomy 8, and trisomy 21 occurred more frequent in patients with inv(16), whereas del(9q) and loss of sex chromosomes were more frequent in t(8;21)-bearing patients with CBF-AML. These data are consistent with data reported in previous studies.11,18-22
The prognostic significance of the secondary chromosome abnormalities in CBF-AML has been investigated in previous studies with somewhat conflicting results. A recent study20 showed that loss of the X chromosome in female patients with t(8;21) predicts a favorable prognosis, whereas neither we nor Schlenk et al21 found significant impact of chromosome X loss on survival of female patients with t(8;21). Loss of the Y chromosome in males with t(8;21) conferred shorter OS and higher relapse risk in some21,22 but not all9,19 studies, including the current one. In contrast to 2 large studies,9,21 which found no association between del(9q) and the patients’ outcome, MVA analysis in our study revealed that t(8;21)-positive patients with del(9q) had a longer OS and a trend toward better DFS. Marcucci et al19 also reported a significantly longer OS in Black, but not in White, patients with t(8;21) and del(9q). A novel contribution of our study is the observation that ploidy levels play a prognostic role in patients with t(8;21), in whom hypodiploidy was favorably associated with DFS, whereas hyperdiploidy was associated with longer OS.
Trisomy 22, the most frequent secondary abnormality in patients with inv(16), has been associated with better outcome in some studies,19,21 but it had no significant impact on survival in another study9 or in ours. The prognostic significance of trisomy 8 has also varied, from being adverse23 to having no significant impact on clinical outcome21 or being associated with a longer survival, which we detected in MVAs in our study, as did Rogers et al.24 Clearly, the role of trisomy 8 in predicting prognosis should be investigated further by large studies. Finally, the presence of other secondary abnormalities (except trisomy 8) was associated in our study with a shorter survival in patients with inv(16).
Interestingly, the presence of ≥3 cytogenetic abnormalities, which is considered to represent a complex karyotype and is generally associated with a poor prognosis in AML types other than CBF-AML,25 was uncommon in our study and was not associated with a worse outcome, confirming previous reports.24,26 Mosna et al14 suggested that a complex karyotype, defined by the presence of ≥4 cytogenetic abnormalities, may be associated with adverse survival. When we used this definition, neither DFS nor OS was significantly worse in patients with a complex karyotype.
AML is a molecularly and cytogenetically diverse disease at initial diagnosis and can relapse through gaining mutations in 1 or more malignant subclones.27 Our study also showed that 21% of the patients with AML who had inv(16) and 23% of the those with t(8;21) had cytogenetic subclones at the time of diagnosis. Despite the high prevalence of this cytogenetic heterogeneity, it has not been considered in outcome analyses so far. After excluding information on the presence or absence of karyotypically normal cells, the presence of subclones was not associated with survival outcomes. However, this may be because the small sample size limited the statistical power needed to detect differences.
The relationship between KIT mutations and an unfavorable outcome was previously established in patients with CBF-AML.28-30 The advent of next-generation sequencing confirms the heterogeneity of AML by molecular profiling and has enabled recognition of KIT mutations outside of hotspots such as codon 816.31 In CBF-AML, the most common KIT mutations are in exon 17 (particularly in adults) and in exon 8 (particularly in children).30,32 KIT mutations confer a poor prognosis in general, but the role of KIT mutations appears to differ between patients within each subset of CBF-AML.30 However, more published data support the notion that KIT mutations are associated with unfavorable outcomes for patients with t(8;21), rather than for those with inv(16).28,33,34 Patients with KIT mutations with CBF-AML may have an associated systemic mastocytosis. We did not examine patients for this purpose specifically in this study, although CBF-AML with associated systemic mastocytosis may have a poorer prognosis, especially when the patients also harbored a high-allelic-burden KIT mutation or the KIT 816 mutation.35
In our study, the DFS of patients with t(8;21) was significantly longer than that of patients with inv(16), although this was not translated into better OS of those with t(8;21). This is not a common finding, another study has shown that relapse, albeit not significantly, may be more common in inv(16),36 perhaps due to patients with t(8;21) AML having significantly lower WBC at diagnosis than patients with inv(16) AML, because WBC at diagnosis is a factor in poor prognosis and a predictor of relapse.3,37 This observation may also be a result of the study population’s heterogeneity; for example, patients with inv(16) were treated in the early years. Although DFS was different, OS was similar in each subset of CBF-AML, indicating that patients with inv(16) can be treated more effectively (eg, achieve a CR2) after relapse. This observation is not unique: a 2005 Cancer and Leukemia Group B study also described a higher cumulative incidence of relapse in inv(16) (57% vs 48%), and, in the same study, a higher DFS was seen in t(8;21) but was not statistically significant at 36 months.19 Other studies have demonstrated similar findings.3,21,38-40 The difference in DFS between each cytogenetic group may also be related to the timing of alloHCT; it was performed more frequently in CR1 in patients with t(8;21). In addition, patients with t(8;21) were diagnosed in later years; therefore, they may have received better management.
Our study has several limitations. The retrospective analysis of data from 12 centers had incomplete or missing data, especially regarding molecular testing. Outside of the KIT D816 mutation, specific variant data for other KIT mutations were not captured and a large subset of patients were seen before 2006 when mutation testing became a standard of care. Some details in treatment were also missing. There was no significant difference between patients who had complete treatment data and those who did not, mainly because treatment in CBF-AML has been standard (successful induction with 7+3 consolidated with high-dose cytarabine) over the past several decades. For the most part, cytogenetic data were available only at diagnosis, which precluded the analysis of clonal evolution at relapse. We had no evaluation of minimum residual disease, which has been reported as an important predictive factor in AML outcome.6,41 In addition, no extensive fluorescence in situ hybridization studies could be performed, and no centralized karyotyping was organized. Finally, our study lacks the next-generation sequencing data that may also have added to prognostication in our patients with CBF-AML.
Despite the limitations, in the large group of patients with a relatively rare subtype of AML, we demonstrated that CBF-AML with inv(16) or t(8;21) are cytogenetically heterogeneous diseases. Ploidy levels, other than pseudodiploidy, trisomy 8, and deletion of 9q, stand out as potentially significant cytogenetic findings with beneficial prognostic implications. In the current molecular era, conventional cytogenetic analysis at diagnosis continues to reveal chromosome abnormalities that affect the prognosis of patients with CBF-AML.
For original data, please contact celalettin_ustun@rush.edu.
Acknowledgments
This work was supported by Austrian Science Fund (FWF) grants F4701-B20 and F4704-B20 (P.V.) and by the ARUP Institute for Clinical and Experimental Pathology (T.I.G.).
Authorship
Contribution: S.y.H., C.C.S.Y., and C.U. conceived the idea, collected and analyzed the data, searched the literature, and wrote and edited the manuscript; K.M. analyzed the cytogenetics data and wrote and edited the manuscript; J.V. and Q.W. performed the statistical analyses; C.D.B., J.K., A.S., E.A.M., J.R.H., H.V., R.O., P.M.K., J.L.H., T.K.K., M.B.M., S.P., N.K.K., D.C., L.B.B., M.D., M.R.L., Y.K., H.-P.H., T.I.G., C.A.Y., V.P., R.N., A.-I.S., G.M., L.S., H.J.D., W.R.S., J.G., G. Hoermann, G. Huls, M.L., D.W., G.B., S.B.-O., C.C., C.A., M.-A.P., P.V., and M.A.L. collected and analyzed the data and wrote and edited the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
The current affiliation for G. Hoermann is MLL Munich Leukemia Laboratory, Munich, Germany.
Clara D. Bloomfield died on 1 March 2020.
Correspondence: Cecilia C. S. Yeung, Fred Hutchinson Cancer Research Center, 1100 Fairview Ave N., Seattle, WA 98109; e-mail: cyeung@fredhutch.org.

